Berikut merupakan kutipan ilmiah Toksikologi-Biokimia kedokteran yang bermanfaat sehingga disusun dan digunakan sebagai referensi pribadi.
Perpustakaan Keluarga : Helmut Todo Tua Simamora/dr. Olga Y.V Hutapea
Cyanide is a rapidly acting lethal agent that is limited in its military usefulness by its high LCt50 and high volatility. Death occurs in 6 to 8 minutes after inhalation of a high Ct. Sodium nitrite and sodium thiosulfate are effective antidotes.
The French used about 4000 tons of cyanide in WWI without notable military success, possibly because the small one- to two-pound munitions used could not deliver the large amounts needed to cause biological effects. Other factors included the high volatility of cyanide (which quickly evaporated and dispersed) and its "all or nothing" biological activity, i.e., it caused few effects below the lethal Ct (this is in contrast to mustard, which causes eye damage at 1% of the lethal amount).
The U.S. maintained a small number of cyanide munitions during World War II (WWII). Japan allegedly used cyanide against China before and during WWII, and Iraq may have used cyanide against the Kurds in the 1980s.
Terms: The term cyanide refers to the anion CN-, or to its acidic form, hydrocyanic acid (HCN). Cyanogen (C2N2) is formed by the oxidation of cyanide ions; however, the term cyanogen has also come to refer to a substance that forms cyanide upon metabolism and produces the biological effects of free cyanide (the term cyanogen is from "cyano" and "gennan," Greek meaning "to produce"). A simple cyanide (HCN, NaCN) is a compound that dissociates to the cyanide anion (CN-) and a cation (H+, Na+). A nitrile is an organic compound that contains cyanide. Acyanogen usually refers to a nitrile that liberates the cyanide anion during metabolism and produces the biological effects of the cyanide anion. Cyanogens may be simple (cyanogen chloride) or complex (sodium nitroprusside).
Cyanides are also called "blood agents," an antiquated term still used by many in the military. At the time of the introduction of cyanide in World War I, the other chemical agents in use caused mainly local effects: riot control agents injured the skin and mucous membranes from direct contact, and phosgene damaged the lungs after inhalation. In contrast, cyanide when inhaled produced systemic effects and was thought to be carried in the blood; hence the term "blood agent." The widespread distribution of absorbed nerve agents and vesicants via the blood invalidates this term as a specific designator for cyanide. Also, the use of "blood agent" for cyanide connotes to some people that the site of action of cyanide is in the blood, an erroneous notion.
Materials of interest as chemical agents are the cyanide hydrogen cyanide (hydrocyanic acid; AC) and the simple cyanogen, cyanogen chloride (CK). Cyanogen bromide was used briefly in World War I, but is of no present interest.
Sources other than military: The cyanide ion is ubiquitous in nearly all living organisms which tolerate and even require the ion in low concentrations. The fruits and seeds (especially pits) of many plants, such as cherries, peaches, almonds, and lima beans, contain cyanogens capable of releasing free cyanide following enzymatic degradation. The edible portion (the roots) of the cassava plant (widely used as a food staple in many parts of the world) is also cyanogenic. The combustion of any material containing carbon and nitrogen has the potential to form cyanide; some plastics (particularly acrylonitriles) predictably release clinically significant amounts when burned. Industrial concerns in the U.S. manufacture over 300,000 tons of hydrogen cyanide annually. Cyanides find widespread use in chemical syntheses, electroplating, mineral extraction, dyeing, printing, photography, and agriculture, and in the manufacture of paper, textiles, and plastics.
The cyanides are in liquid state in munitions, but rapidly vaporize upon detonation of the munitions. The major threat is from the vapor. The liquid toxicity is about that of mustard (see toxicity, below).
The preferred way to deliver cyanide is by large munitions (bombs, large shells), because smaller weapons will not provide the concentrations needed for effects.
Cyanide has a high affinity for certain sulfur compounds (sulfanes, which contain two covalently bonded but unequally charged sulfur atoms) and for certain metallic complexes, particularly those containing cobalt and the trivalent form of iron (Fe3+). The cyanide ion can rapidly combine with iron in cytochrome a3 (a component of the cytochrome aa3 or cytochrome oxidase complex in mitochrondria) to inhibit this enzyme, thus preventing intracellular oxygen utilization. The cell then utilizes anaerobic metabolism, creating excess lactic acid and a metabolic acidosis. Cyanide also has a high affinity for the ferric iron of methemoglobin and one therapeutic strategem induces the formation of methemoglobin to which cyanide preferentially binds.
The small quantity of cyanide always present in human tissues is metabolized at the approximate rate of 17 Fg/kg"min, primarily by the hepatic enzyme rhodanese, which catalyzes the irreversible reaction of cyanide and a sulfane to produce thiocyanate, a relatively nontoxic compound excreted in the urine. (An elevated concentration of thiocyanate in either blood or urine is evidence of cyanide exposure.) The limiting factor under normal conditions is the availability of a sulfane as a substrate for rhodanese, and sulfur is administered therapeutically as sodium thiosulfate to accelerate this reaction. Because of the ability of the body to detoxify small amounts of cyanide via the rhodanese-catalyzed reaction with sulfane, the lethal dose of cyanide is time dependent; that is, a given amount of cyanide absorbed slowly may cause no biological effects even though the same amount administered over a very short period of time may be lethal. In contrast, the LCt50 of each of the other chemical agents, which are not metabolized to the same extent as is cyanide, is relatively constant over time, and a lethal amount causes death whether administered within minutes or over hours.
Toxicities: Cyanide is the least toxic of the "lethal" chemical agents. The LCt50s of AC and CK by inhalation have been estimated to be 2500-5000 mg"min/m3 for AC and about 11,000 mg"min/m3 for CK. LD50s for hydrogen cyanide have been estimated to be 1.1 mg/kg for intravenous administration and 100 mg/kg after skin exposure. The oral LD50s for sodium and potassium cyanide are about 100 and 200 mg/kg respectively.
Cyanide is unique among military chemical agents because it is detoxified at a rate that is of practical importance, about 17 Fg/kg"min. As a result the LCt50 is greater for a long exposure (e.g., 60 min) than for a short exposure (e.g., 2 min).
Effects: The organs most susceptible to cyanide are the central nervous system (CNS) and the heart. Most clinical effects are of CNS origin and are nonspecific.
About 15 seconds after inhalation of a high concentration of cyanide vapor concentration there is a transient hyperpnea followed in 15-30 seconds by the onset of convulsions. Respiratory activity stops two to three minutes later, and cardiac activity ceases several minutes later still, or at about six to eight minutes after exposure.
The onset and progression of signs and symptoms after ingestion of cyanide or after inhalation of a lower concentration of vapor are slower. The first effects may not occur until several minutes after exposure, and the time course of these effects depends on the amount absorbed and the rate of absorption. The initial transient hyperpnea may be followed by a feelings of anxiety or apprehension, agitation, vertigo, a feeling of weakness, nausea with or without vomiting, and muscular trembling. Later, consciousness is lost, respiration decreases in rate and depth, and convulsions, apnea, and cardiac dysrhythmias and standstill follow. Because this cascade of events is prolonged, diagnosis and successful treatment are possible.
The effects of cyanogen chloride include those described for hydrogen cyanide. Cyanogen chloride is also similar to the riot control agents in causing irritation to the eyes, nose, and airways as well as marked lacrimation, rhinorrhea, and bronchosecretions.
Physical Findings: Physical findings are few and non-specific. The two that are said to be characteristic are in fact not always observed. The first is severe respiratory distress in an acyanotic individual. When seen, "cherry-red" skin suggests either circulating carboxyhemoglobin from carbon monoxide poisoning or a high venous oxygen content from failure of extraction of oxygen by tissues poisoned by cyanide or hydrogen sulfide. However, cyanide victims may have normal appearing skin and may even be cyanotic, although cyanosis is not classically associated with cyanide poisoning.
Table: Cyanide (AC and CK)
Effects from vapor exposure
| Moderate, from low concentration | Transient increase in rate and depth of breathing, dizziness,nausea, vomiting, headache | These may progress to severe effects if exposure continues. | The time of onset of these effects depends on the concentration, but is often within minutes after start of exposure. |
| Severe, from high concentration | Transient increase in rate and depth of breathing -- 15 secondsConvulsions -- 30 seconds Cessation of respiration -- 2-4 minutes Cessation of heartbeat -- 4-8 minutes |
In addition to the above, CK causes intense irritation of the eyes, nose, and airways.
The second classic sign is the odor of bitter almonds. However, about 50% of the population is genetically unable to detect the odor of cyanide.
The casualty may be diaphoretic with normal sized or large pupils. An initial hypertension and compensatory bradycardia are followed by a declining blood pressure and tachycardia. Terminal hypotension is accompanied by bradyarrhythmias before asystole.
Effects begin in 15 seconds following inhalation of a lethal Ct; death ensues in six to eight minutes. The onset of effects following inhalation of lower Cts may be as early as minutes after the beginning of the exposure. After exposure is terminated by evacuation to fresh air or by masking, there is little danger of delayed onset of effects.
Battlefield inhalational exposure to either cyanide or a nerve agent may precipitate the sudden onset of loss of consciousness followed by convulsions and apnea. The nerve agent casualty has miosis (until shortly before death), copious oral and nasal secretions, and muscular fasciculations. The cyanide casualty has normal sized or dilated pupils, few secretions, and muscular twitching but no fasciculations. In addition, the nerve agent casualty may be cyanotic, and the cyanide casualty usually is not cyanotic.
1. An elevated blood cyanide concentration: Mild effects may be apparent at concentrations of 0.5-1.0 Fg/mL, and concentrations of 2.5 Fg/mL and higher are associated with coma, convulsions and death.
2. Acidosis: Metabolic acidosis with a high concentration of lactic acid (lactic acidosis), or a metabolic acidosis with an unexplained high anion gap (if the means to measure lactic acid are not available) may be present. Because oxygen cannot be utilized, anaerobic metabolism with the production of lactic acid replaces aerobic metabolism. Lactic acidosis, however, may reflect other disease states and is not specific for cyanide poisoning.
3. Oxygen content of venous blood greater than normal. This also is because of poisoning of the intramitochondrial respiratory chain and the resulting failure of cells to extract oxygen from arterial blood. This finding is also not specific for cyanide poisoning.
The primary goal in therapy is to remove the cyanide from the enzyme cytochrome a3 in the cytochrome oxidase complex. A complicating factor is the rapidity with which cyanide, particularly inhaled cyanide, causes death.
A secondary goal is to detoxify or bind the cyanide so that it can not reenter the cell to reinhibit the enzyme. A closely associated goal is supportive management.
Methemoglobin has a high affinity for cyanide, and cyanide will preferentially bind to methemoglobin rather than to the cytochrome. Most methemoglobin formers have clinically significant side effects. The nitrites, which were first used to antagonize the effects of cyanide over a century ago, cause orthostatic hypotension, but this is relatively insignificant in a supine casualty. Amyl nitrite, historically the first nitrite used, is an volatile substance formulated in a perle that is crushed or broken for the victim to inhale. In an apneic patient a means of ventilation is necessary.
Another methemoglobin former, sodium nitrite, is formulated for intravenous use. The standard ampule contains 300 mg of the drug in 10 mL of diluent, and this is injected intravenously over a two- to four-minute period.
Detoxification (metabolism) of cyanide is accomplished by the administration of a sulfur-containing compound that combines with cyanide to produce thiocyanate, a relatively non-toxic substance which is rapidly excreted via the kidneys. The hepatic enzyme rhodanese catalyzes the one-way reaction of cyanide and a sulfane to thiocyanate. Sodium thiosulfate is packaged in a 50-mL ampule containing 12.5 grams of the drug. Intravenous injection of all 12.5 grams follows successful completion of the intravenous injection of sodium nitrite. Half of the original dosage of each drug may be repeated if symptoms persist.
An immediate casualty is one who presents within minutes of inhalational exposure with convulsions or the recent onset of apnea, but with circulation intact. Immediate antidote administration will be lifesaving.
A minimal casualty is one who has inhaled less than a lethal amount and has mild effects. The antidotes may reduce his symptoms, but are not lifesaving.
The delayed casualty is one recovering from mild effects or from successful therapy. Generally, it will be hours before full recovery. Evacuation is not necessary, but might be considered until full recovery takes place.
An expectant casualty is apneic with circulatory failure.
Generally, a casualty who has had inhalation exposure and survives long enough to reach medical care will need little care.
Full recovery is usually relatively fast after cyanide intoxication. Those with mild to moderate effects from the agent can usually return to duty within hours, and those successfully treated after severe effects can return within a day.
Approach Considerations
The workup in patients with cyanide exposure may include the studies discussed below.
Arterial and venous blood gases
Cyanide toxicity is characterized by a normal arterial oxygen tension and an abnormally high venous oxygen tension, resulting in a decreased arteriovenous oxygen difference (< 10%). A high-anion-gap metabolic acidosis is a hallmark of significant cyanide toxicity.[7, 19] Apnea may result in combined metabolic and respiratory acidosis.
Blood lactate level
Elevation in the blood lactate level is a sensitive marker for cyanide toxicity. A plasma lactate concentration of greater than 10 mmol/L in smoke inhalation or greater than 6 mmol/L after reported or strongly suspected pure cyanide poisoning suggests significant cyanide exposure.[20]
Red blood cell or plasma cyanide concentration
Cyanide blood concentrations are not generally available in time to aid in the treatment of acute poisoning, but may provide subsequent confirmation. In cyanogen exposures, these tests provide documentation for therapeutic use, which may last several days.
The preferred test is a red blood cell cyanide concentration. With this method, mild toxicity is observed at concentrations of 0.5-1.0 μg/mL. Concentrations of 2.5 μg/mL and higher are associated with coma, seizures, and death. Blood cyanide concentrations may artificially increase after sodium nitrite (antidote) administration, because of in vitro release of cyanide from cyanomethemoglobin during the analytical procedure by strong acid used in analysis.
Carboxyhemoglobin level or blood carbon monoxide concentration
Carboxyhemoglobin (HbCO) level (by co-oximetry) or blood carbon monoxide concentration (by infrared spectroscopy) may be obtained in patients with smoke inhalation to rule out concurrent exposure. HbCO measurements may be artificially elevated in blood samples drawn after hydroxocobalamin administration.[21]
Methemoglobin level
A methemoglobin level is especially important in cyanotic patients. The presence of methemoglobin suggests that little or no free cyanide is available for binding, because methemoglobin vigorously binds cyanide to form cyanomethemoglobin (which is not measured as methemoglobin).
Methemoglobin concentrations provide a guide for continued therapy after the use of methemoglobin-inducing antidotes, such as sodium nitrite. Elevated levels of methemoglobin (>10%) indicate that further nitrite therapy is not indicated and, in fact, may be dangerous.
Electrocardiogram (ECG)
On ECG, nonspecific findings predominate. Abnormalities may include the following[22] :
- Sinus bradycardia or tachycardia
- Atrioventricular blocks
- Supraventricular or ventricular arrhythmias
- Ischemic electrocardiographic changes
In some cases, shortening of the ST segment with eventual fusion of the T wave into the QRS complex has been observed.
Other
No imaging studies are indicated acutely for cyanide exposure, but magnetic resonance imaging (MRI) may be useful during the evaluation of postexposure neurologic sequelae.
Fluorescein staining and slit-lamp examination of the eyes may be necessary following decontamination to assess corneal integrity.
Cyanides comprise a wide range of compounds of varying degrees of chemical complexity, all of which contain a CN moiety, to which humans are exposed in gas, liquid, and solid form from a broad range of natural and anthropogenic sources. While many chemical forms of cyanide are used in industrial application or are present in the environment, the cyanide anion CN– is the primary toxic agent, regardless of origin.
Hydrogen cyanide is a colourless or pale blue liquid or gas with a faint bitter almond-like odour. Hydrogen cyanide is used primarily in the production of substances such as adiponitrile, methyl methacrylate, chelating agents, cyanuric chloride, methionine and its hydroxylated analogues, and sodium and potassium cyanide. Hydrogen cyanide is also used as a fumigant in ships, railroad cars, large buildings, grain silos, and flour mills, as well as in the fumigation of peas and seeds in vacuum chambers.
Other cyanides, such as sodium and potassium cyanide, are solid or crystalline hygroscopic salts widely used in ore extracting processes for the recovery of gold and silver, electroplating, case-hardening of steel, base metal flotation, metal degreasing, dyeing, printing, and photography. They are also widely used in the synthesis of organic and inorganic chemicals (e.g., nitriles, carboxylic acids, amides, esters, and amines; heavy metal cyanides) and in the production of chelating agents.
Anthropogenic sources of cyanide release to the environment are diverse. Releases to air include chemical manufacturing and processing industries, such as metallurgical industries and metal plating, and extraction of gold and silver from low-grade ores. Other sources include volatilization from cyanide wastes disposed of in landfills and waste ponds, emissions from municipal solid waste incinerators, biomass burning, fossil fuel combustion, including vehicle emissions, fumigation operations, and the production of coke or other coal carbonization procedures.
Hydrogen cyanide is formed during the incomplete combustion of nitrogen-containing polymers, such as certain plastics, polyurethanes, and wool. Hydrogen cyanide is present in cigarette smoke.
Non-point sources of cyanide released to water can result from runoff from cyanide-containing anti-caking salts used on roads, migration from landfills, and agricultural and atmospheric fallout and washout. Point sources of releases to water include discharges from gold mining plants, wastewater treatment works, iron and steel production, and organic chemical industries.
Principal natural sources of cyanides are over 2000 plant species, including fruits and vegetables, that contain cyanogenic glycosides, which can release cyanide on hydrolysis when ingested. Among them, cassava (tapioca, manioc) and sorghum are staple foods for hundreds of millions of people in many tropical countries. Known cyanogenic glycosides in plants include amygdalin, linamarin, prunasin, dhurrin, lotaustralin, and taxiphyllin. Hydrogen cyanide is released into the atmosphere from natural biogenic processes from higher plants, bacteria, and fungi.
In air, cyanide is present as gaseous hydrogen cyanide, with a small amount present in fine dust particles. Cyanides have the potential to be transported over long distances from their respective emission sources.
The majority of the population is exposed to very low levels of cyanide in the general environment. There are, however, specific subgroups with higher potential for exposure. These include individuals involved in large-scale processing of cassava and those consuming significant quantities of improperly prepared foods containing cyanogenic glycosides, such as cassava, speciality foods such as apricot pits, and bitter almonds. Other subgroups with greatest potential for exposure include those in the vicinity of accidental or intended releases from point sources, active and passive smokers, and fire-related smoke inhalation victims.
Workers may be exposed to cyanides during fumigation operations and the production and use of cyanides in many industrial processes — for example, electroplating, case-hardening of steel, and extraction of gold and silver from ores.
Cyanides are well absorbed via the gastrointestinal tract or skin and rapidly absorbed via the respiratory tract. Once absorbed, cyanide is rapidly and ubiquitously distributed throughout the body, although the highest levels are typically found in the liver, lungs, blood, and brain. There is no accumulation of cyanide in the blood or tissues following chronic or repeated exposure.
Approximately 80% of absorbed cyanide is metabolized to thiocyanate in the liver by the mitochondrial sulfur transferase enzyme rhodanese and other sulfur transferases. Thiocyanate is excreted in the urine. Minor pathways for cyanide detoxification involve reaction with cystine to produce aminothiazoline- and iminothiazolidinecarboxylic acids and combination with hydroxycobalamin (vitamin B12a) to form cyanocobalamin (vitamin B12); these end-products are also excreted in the urine.
The principal features of the toxicity profile for cyanide are its high acute toxicity by all routes of administration, with a very steep and rate-dependent dose–effect curve, and chronic toxicity, probably mediated through the main metabolite and detoxification product, thiocyanate. The toxic effects of cyanide ion in humans and animals are generally similar and are believed to result from inactivation of cytochrome oxidase and inhibition of cellular respiration and consequent histotoxic anoxia. The primary targets of cyanide toxicity in humans and animals are the cardiovascular, respiratory, and central nervous systems. The endocrine system is also a potential target for long-term toxicity, as a function of continued exposure to thiocyanate, which prevents the uptake of iodine in the thyroid and acts as a goitrogenic agent.
In humans, whereas slight effects occur at exposure levels of 20–40 mg/m3, 50–60 mg/m3 can be tolerated without immediate or late effects for 20 min to 1 h, 120–150 mg/m3 may lead to death after 0.5–1 h, 150 mg/m3 is likely to be fatal within 30 min, 200 mg/m3 is likely fatal after 10 min, and 300 mg/m3 is immediately fatal. The lowest reported oral lethal dose for humans is 0.54 mg/kg body weight, and the average absorbed dose at the time of death has been estimated at 1.4 mg/kg body weight (calculated as hydrogen cyanide). Sequelae after severe acute intoxications may include neuropsychiatric manifestations and Parkinson-type disease. Cyanide from tobacco smoke has been implicated as a contributing factor in tobacco–alcohol amblyopia. Long-term exposure to lower concentrations of cyanide in occupational settings can result in a variety of symptoms related to central nervous system effects.
Long-term consumption of cassava containing high levels of cyanogenic glycosides has been associated with tropical ataxic neuropathy, spastic paraparesis, and, in areas with low iodine intake, development of hypothyroidism, goitre, and cretinism. While exposure to cyanide has been crudely estimated to be 15–50 mg/day in endemic areas in some such cases, owing to the limitations of data on exposure and potential impact of confounders such as malnutrition, low protein content of the diet, vitamin deficiencies, and iodine status, the available data do not provide meaningful information on dose–response for cyanide.
Data on end-points other than acute toxicity are somewhat limited. This is attributable in large part to difficulties in conducting, for example, investigations of repeated-dose or chronic toxicity due to the high acute toxicity of the compound. Cyanides are weakly irritating to the skin and eye; data on sensitizing properties or carcinogenicity of hydrogen cyanide or its alkali salts have not been identified. Although somewhat limited, the weight of evidence of available data indicates that cyanide is not genotoxic and that it induces developmental effects only at doses or concentrations that are overtly toxic to the mothers.
Available data in human populations are considered inadequate as a basis for characterization of dose–response for chronic ingestion of cyanide. In a 13-week repeated-dose toxicity study in which cyanide was administered in drinking-water, there were no clinical signs associated with central nervous system effects or histopathological effects in the brain or thyroid of rats or mice exposed to doses up to 12.5 mg and 26 mg cyanide/kg body weight per day, respectively. At 12.5 mg cyanide/kg body weight per day, there were slight changes in the reproductive tract in male rats, which, although they apparently would not affect fertility in rats, are possibly significant to humans. The no-observed-adverse-effect level (NOAEL) for these effects was 4.5 mg/kg body weight per day. The examination of neurotoxicity in this study was limited to clinical observation and optical microscopy in autopsy. The few available studies specifically intended to investigate neurotoxicity, while reporting adverse effects at exposure levels of 1.2 mg cyanide/kg body weight per day in rats and 0.48 mg cyanide/kg body weight per day in goats, suffer from weaknesses that preclude their quantitative assessment.
In relation to characterization of concentration–response for repeated-dose toxicity for inhalation (relevant principally to the occupational environment), in three separate studies in rats, there were no adverse systemic effects in rats exposed to acetone cyanohydrin, which is rapidly hydrolysed to hydrogen cyanide at physiological pH, at concentrations up to 211 mg/m3 (corresponding to a concentration of 67 mg hydrogen cyanide/m3). The steepness of the dose–effect curve is illustrated by the observation of 30% mortality among rats exposed part of the day to 225 mg acetone cyanohydrin/m3 (71 mg hydrogen cyanide/m3).
Adverse effects of exposure to the low concentrations of cyanide that are generally present in the general environment (<1 µg/m3 in ambient air; <10 µg/litre in water) are unlikely. Acute cyanide intoxications may arise from eating apricot kernels, choke cherries, and other stone fruit kernels with high concentrations of cyanogenic glycosides. Inadequately prepared cassava, when constituting the major part of the diet, may be hazardous.
Hydrogen cyanide (HCN) is a colourless or pale blue liquid or gas with a faint bitter almond-like odour. Common synonyms are hydrocyanic acid and prussic acid. Hydrogen cyanide is a very weak acid, with a pKa value of 9.22 at 25 °C. It is soluble in water and alcohol. Hydrogen cyanide is commercially available as a gas or as a technical-grade liquid in concentrations of 5, 10, and 96–99.5%. Phosphoric acid is added to liquid hydrogen cyanide as a stabilizer to prevent decomposition and explosion (ATSDR, 1997). Some important physical and chemical properties of hydrogen cyanide are summarized in Table 1.
The conversion factors2 for hydrogen cyanide in air (at 20 °C and 101.3 kPa) are as follows:
1 ppm = 1.12 mg/m3
1 mg/m3 = 0.890 ppm
Table 1: Physical and chemical properties of hydrogen cyanide (CAS No. 74-90-8 ).a
| Property | Value |
| Relative molecular mass | 27.03 |
| Boiling point (°C) | 25.70 |
| Solubility (30 °C) | Miscible with water; soluble in ethanol |
| Specific density: vapours (31 °C) | 0.937 |
| Odour threshold | 0.7 mg/m3 in air 0.17 mg/litre in water |
| Henry’s law constant (dimensionless) | 180–300b |
| Octanol/water partition coefficient (logKow) | 0.66 |
| Vapour pressure (kPa) | 35.2 at 0 °C 107.2 at 27.2 °C |
a From ACGIH (2001); DECOS (2002).
b Hine & Weimar (1965); Edwards et al. (1978); Gaffney et al. (1987).
Sodium cyanide (NaCN) is a white hygroscopic crystalline powder with a faint bitter almond-like odour. Common synonyms are cyanide of sodium and hydrocyanic acid, sodium. Commercially available sodium cyanide generally achieves a purity of 95–98%. The aqueous solution of sodium cyanide is strongly alkaline and rapidly decomposes. Sodium cyanide produces hydrogen cyanide on contact with acids or acid salts.
Potassium cyanide (KCN) is a white deliquescent solid with an odour of hydrogen cyanide. Common synonyms are hydrocyanic acid, potassium salt and cyanide of potassium. Potassium cyanide is commercially available at a 95% purity. An aqueous solution of potassium cyanide in water is strongly alkaline. Potassium cyanide also produces hydrogen cyanide on contact with acids or acid salts.
Calcium cyanide (Ca(CN)2), also commonly called cyanide of calcium, calcid, or calsyan, is a white crystalline solid. Its aqueous solution gradually liberates hydrogen cyanide. Cyanides such as sodium cyanide, potassium cyanide, and calcium cyanide form strong complexes with many metals (Table 2).
Cyanogen is a colourless toxic gas with an almond-like odour. Common synonyms are carbon nitrile, dicyanogen, ethane dinitrile, and oxalic acid dinitrile. Cyanogen is slowly hydrolysed in aqueous solution, yielding oxalic acid and ammonia. The conversion factors for cyanogen in air at 20 °C and 101.3 kPa are as follows:
1 ppm = 2.16 mg/m3
1 mg/m3 = 0.462 ppm
Table 2: Physical and chemical properties of selected cyanide compounds.a
| Species | CAS number | Molecular formula | Relative molecular mass | Common synonym(s) | Boiling point (°C) | Solubility |
| Sodium cyanide | NaCN | 49.02 | Cyanide of sodium | Soluble in water, slightly soluble in alcohol | ||
| Potassium cyanide | KCN | 65.11 | Cyanide of potassium | Soluble in water, slightly soluble in alcohol | ||
| Calcium cyanide | Ca(CN)2 | 92.12 | Calcid; calsyan | Soluble in water, slightly soluble in alcohol | ||
| Copper cyanide | 54-92-3 | CuCN | 89.56 | Cupricin | Insoluble in water | |
| Potassium silver cyanide | 501-61-6 | KAg(CN)2 | 198.01 | Potassium dicyanoargentate | Soluble in water, slightly soluble in ether | |
| Sodium ferrocyanide | Na4Fe(CN)6 | 303.91 | Sodium hexacyanoferrate (II) | Soluble in water | ||
| Potassium ferrocyanide | 13943-57-3 | K4Fe(CN)6 | 368.35 | Yellow prussiate of potash | Soluble in water | |
| Potassium ferricyanide | K3Fe(CN)6 | 329.95 | Red prussiate of potash | Slowly soluble in 2.5 parts of cold water; decomposes slowly in water | ||
| Cyanogen | NCCN | 52.04 | Carbon nitrile; dicyanogen | –20.7 | Soluble in water, alcohol, and ether | |
| Cyanogen chloride | CNCl | 61.47 | Chlorine cyanide | 13.8 | Soluble in water and alcohol | |
| Acetone cyanohydrin | (CH3)2C(OH)CN | 85.10 | ACH; methyllactonitrile | 82 | Soluble in water | |
| Sodium nitroprusside | Na2[Fe(CN)5NO] | 261.97 | Sodium nitroferrocyanide; sodium nitrosyl pentacyanoferrate (III) | Soluble in 2.3 parts of water, slightly soluble in alcohol |
a From Windholz (1983); ACGIH (2001); ECETOC (2004).
Cyanogen chloride is a colourless gas. Its common synonym is chlorine cyanide, and its common trade name is Caswell No. 267. Cyanogen chloride releases hydrogen cyanide by hydrolysis. Its conversion factors in air are:
1 ppm = 2.56 mg/m3
1 mg/m3 = 0.391 ppm
Common synonyms of acetone cyanohydrin are ACH, 2-cyano-2-propanol, 2-methyllactonitrile, and 2-hydroxy-2-methyl propanenitrile. It dissociates on standing to liberate hydrogen cyanide. Its boiling point is 120 °C (with decomposition to hydrogen cyanide and acetone). Its conversion factors in air are:
1 ppm = 3.54 mg/m3
1 mg/m3 = 0.283 ppm
The half-time of ACH in water was reported to be 9 min (Ellington et al., 1986); further studies reported that this hydrolysis to acetone and hydrogen cyanide was pH dependent, and half-times of 58, 27, and 8 min were observed at pH 4.8, 6.3, and 6.8 (ICI, 1993). In a more recent study, similar findings were reported (half-times of 54.7, 31.2, 5.4, and 4.0 min at pH 6.00, 6.40, 6.86, and 7.00, respectively) (Frank et al., 2002).
Some chemical properties of other cyanides are given in Table 2. Copper cyanide is a white to cream-coloured solid. Its common name is cuprous cyanide, and its synonym is cupricin. Potassium silver cyanide occurs as white crystals; its common synonym is potassium dicyanoargentate. It is sensitive to light. Sodium ferrocyanide decomposes at 435 °C, forming sodium cyanide.
Cyanogenic glycosides are produced naturally by many plants; when hydrolysed, they produce hydrogen cyanide. Chemical structures of some commonly occurring cyanogenic glycosides are depicted in Figure 1.
Further chemical and physical properties of hydrogen cyanide and some cyanides are summarized in the International Chemical Safety Cards included in this document.
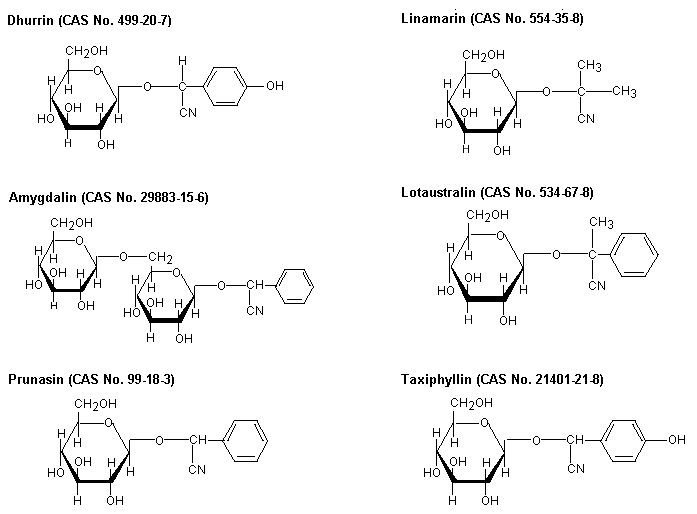
Fig. 1: Cyanogenic glycosides in major edible plants (JECFA, 1993)
Amygdalin occurs in (among others) almonds, dhurrin in sorghum, linamarin in cassava, lotaustralin in cassava and lima beans, prunasin in stone fruits, and taxiphyllin in bamboo shoots.
Cyanides in environmental media are usually collected in sodium or potassium hydroxide solution and measured by spectrophotometry (Agrawal et al., 1991), colorimetry, or ion-specific electrode or by headspace gas chromatography with a nitrogen-specific detector or electron capture detector (Maseda et al., 1989; Seto et al, 1993). Cyanide in aqueous matrices is usually measured by colorimetric, titrimetric (US EPA, 1983), or electrochemical methods after pretreatment to produce hydrogen cyanide and absorption in sodium hydroxide solution. Total cyanide (irrespective of origin) includes all of the available cyanide in a sample; in drinking-water, it is measured by semi-automated colorimetry (EPA Method 335.4) as well as by selective electrode, ultraviolet/distillation/spectrophotometry, and ion chromatography (EPA Method 300.0) (US EPA, 1993a). Free cyanide can also be determined by one method (SM-4500-CN-F) approved for drinking-water compliance monitoring analysis that does not require distillation, the specific ion electrode method (US EPA, 2003a). Weak acid dissociable cyanide analysis (used principally by the precious metals mining industry) includes those cyanide species liberated at moderate pH 4.5, such as aqueous hydrogen cyanide and cyanide anion, the majority of copper, cadmium, nickel, zinc, silver, and tin complexes, and others with similar low dissociation constants. Weak acid dissociable cyanide can be determined in wastewaters by a ligand exchange/flow injection/amperometric technique (EPA Method 1677) (Milosavlievic et al., 1995; US EPA, 1997).
A chromatographic technique with fluorescence detection is used to detect trace amounts of cyanide in blood cells (Chinaka et al., 1998). Cyanide in biological tissue and fluids can be measured spectrophotometrically after reaction with methaemoglobin.
Since many cyanides are unstable and emit volatile hydrogen cyanide gas, sampling, storage, and analysis must be done with caution, preferably immediately upon collection.
The three commonly used techniques (colorimetric, titrimetric, and electrochemical) may all suffer from interference problems, unless proper precautions are taken (ATSDR, 1989).
Metals suppress the transformation of cyanide to formic acid, thus lowering the measured hydrogen cyanide concentration (Dolzine et al., 1982). Carbonyl compounds also decrease the hydrogen cyanide recovery (Honig et al., 1983), as in the case with soybean samples, in which carbonyl compounds occur naturally.
Sodium thiosulfate can interfere with potentiometric (Sylvester et al., 1982) or colorimetric analysis (Ganjeloo et al., 1980). Care should be taken, since it is often used as an antidote to treat chemical poisoning.
Continuous monitoring of cyanide is also available using equipment based on diffusion and amperometric detection of hydrogen cyanide (NIOSH, 1976).
Detection limits for the different methods for hydrogen cyanide range from 0.8 to 400 mg/m3 for air samples, from 0.04 to 200 µg/litre for aqueous samples, and from 0.8 to 300 µg/litre for biological samples. NIOSH Method 7904 for workplace air has a limit of detection of 2.5 µg cyanide (NIOSH, 1994).
Hydrogen cyanide is ubiquitous in nature. It is found in the stratosphere and non-urban troposphere (US EPA, 1990). It is released into the atmosphere from biomass burning, volcanoes, and natural biogenic processes from higher plants, bacteria, algae, and fungi (Fiksel et al., 1981; Cicerone & Zellner, 1983; Way, 1984; ATSDR, 1997; Li et al., 2000). An estimate of the amount of cyanide released to the environment from natural biogenic processes is not available (ATSDR, 1997).
Cyanide occurs naturally as cyanogenic glycosides in at least 2000 plants (Figure 1). Amygdalin (d-mandelonitrile-beta-d-glucoside-6-beta-d-glucoside) has been found in about 1000 species of plants, including cassava (tapioca, manioc), sweet potato, corn, cabbage, linseed, millet, and bamboo, in pits of stone fruits, such as cherries, peaches, and apricots, and in apple seeds (JECFA, 1993; Sharma, 1993; Padmaja, 1995). It is also present in bitter almonds and American white lima beans (Ermans et al., 1972). After ingestion, linamarin can be hydrolysed by either cassava linamarase or an endogenous beta-glucosidase to yield d-glucose and ACH (Frakes et al., 1986a).
Hydrogen cyanide is principally produced by two synthetic catalytic processes involving the reaction of ammonia and natural gas (or methane) with or without air. It is also obtained as a by-product in the production of acrylonitrile by the ammoxidation of propylene, which accounts for approximately 30% of the worldwide production of hydrogen cyanide.
Sodium and potassium cyanides are principally prepared by the direct reaction of hydrogen cyanide with the respective alkali in closed systems (European Chemicals Bureau, 2000a,b). Sodium cyanide is also prepared to a lesser extent by melting sodium chloride with calcium cyanamide or by heating sodium amide salt with carbon.
Calcium cyanide is produced by the reaction of coke, coal, and limestone.
Cyanogen chloride is a reaction product of organic precursors with hypochlorous acid in the presence of ammonia and may be formed as a by-product of the chloramination of water (WHO, 1996; IPCS, 2000a).
ACH was first produced in the 1930s as an intermediate in the production of methyl methacrylate from hydrogen cyanide. It is currently produced from the liquid-phase reaction of hydrogen cyanide and acetone in the presence of an alkali catalyst at atmospheric pressure (ECETOC, 2004).
Hydrogen cyanide capacity is generally treated as the sum of purposeful direct synthesis and that derived as a by-product of acrylonitrile production. Annual US hydrogen cyanide capacity by 11 companies in 1991 was 666 000 tonnes. US production of hydrogen cyanide from 1983 to 1989 rose from 300 000 to 445 000 tonnes (Pesce, 1993). Output of hydrogen cyanide in the USA was 545 000 tonnes in 1992 (Cohrssen, 2001). Worldwide annual production and capacity of hydrogen cyanide in 1992 were estimated to be 950 000 and 1 320 000 tonnes, respectively (Pesce, 1993; Cohrssen, 2001). It has been estimated that the present total annual production of hydrogen cyanide worldwide is 1.4 million tonnes (Mudder & Botz, 2000).
In 1983, the major end uses of hydrogen cyanide in the USA were in the production of adiponitrile (200 000 tonnes), ACH (128 000 tonnes), cyanuric chloride (28 500 tonnes), sodium cyanide (69 000 tonnes), chelating agents (15 800 tonnes), and nitrilotriacetic acid (10 100 tonnes) and for miscellaneous uses (20 000 tonnes) (US EPA, 1990). Hydrogen cyanide is also used in the production of methyl methacrylate, methionine and its hydroxylated analogues, and potassium cyanide (ATSDR, 1997; ECETOC, 2004).
Sodium cyanide is extensively employed in a large number of industrial processes, including electroplating and case-hardening of metals; the extraction (cyanidation) of gold and silver from ores; base metal flotation; coal gasification; and the fumigation of ships, railroad cars, buildings, grain silos, flour mills, seeds in vacuum chambers, and soil. Large quantities of sodium cyanide are used to introduce cyano groups into organic compounds, in particular through a reaction with organic halogen compounds to yield nitriles. The nitriles can then be converted to a variety of carboxylic acids, amides, esters, and amines. Potassium cyanide is used for electrolytic refining of platinum, for metal colouring, and as an electrolyte for the separation of gold, silver, and copper from platinum (Eisler et al., 1999; Patnaik, 1999; ACGIH, 2001; ECETOC, 2004). Cyanide salts are used as chelating agents, and the complex cyanides of copper, zinc, and cadmium are used in electroplating processes, principally the plating of iron, steel, and zinc (ECETOC, 2004).
Calcium cyanide is used chiefly as a fumigant, because it readily releases hydrogen cyanide when exposed to air; as a fertilizer, defoliant, herbicide, and rodenticide; as a stabilizer for cement; and in stainless steel manufacture (ACGIH, 2001).
Cyanogen is used as a fumigant, as a fuel gas for welding and cutting heat-resistant metals, and as a rocket and missile propellant (ATSDR, 1997).
Cyanogen chloride is used as a fumigant gas and as a reagent in chemical synthesis.
Cuprous cyanide is used in plating baths for silver, brass, and copper–tin alloy plating (ATSDR, 1997), as an antifouling agent in marine paint, and as an insecticide and fungicide (Windholz, 1983).
Potassium silver cyanide is used in silver plating and as a bactericide.
Potassium ferricyanide is used chiefly for blueprints, in photography, for staining wood, in calico printing, and in electroplating.
Sodium ferrocyanide is used in ore flotation, as an anti-caking agent in rock salt, and in photography for bleaching, toning, and fixing.
Sodium nitroprusside has been used as an antihypertensive agent and in congestive heart failure and is used for deliberate induction of hypotension during certain neurosurgical procedures.
ACH is used in preparative transcyanohydrination reactions.
More than 30 large-scale accidental releases of cyanide to water systems have been reported since 1975; these include transportation accidents, pipe failures, and tailings dam-related releases (Korte et al., 2000; Mudder & Botz, 2000).
Non-point sources of cyanide released to water can result from runoff from cyanide-containing anti-caking salts (i.e., sodium ferrocyanide) used on roads, migration from landfills, and agricultural and atmospheric fallout and washout (ATSDR, 1997).
The extraction of gold from low-grade ores by cyanidation processes was estimated to result in a worldwide emission of 20 000 tonnes of hydrogen cyanide into the atmosphere (Korte & Coulston, 1998). Another estimate suggested that currently 45 300 tonnes of cyanide are used in the USA in the cyanidation process. The wastes from these processes result in large cyanide-containing ponds near the mining operations (Clark & Hothem, 1991; Henny et al., 1994; Ma & Pritsos, 1997; Eisler et al., 1999).
The major point sources of cyanide release to water are discharges from gold mining plants, publicly owned wastewater treatment plants, iron and steel production, and the organic chemical industries. An estimated 3 billion litres (i.e., 3 × 109 litres) of wastes containing cyanides were generated in the USA in 1983, principally from spent cyanide plating bath solutions from electroplating operations (except for precious metals) and from spent stripping and cleaning bath solutions from electroplating operations (Grosse, 1986).
During cassava starch production, large amounts of cyanoglycosides are released and hydrolysed by plant-borne enzymes, leading to cyanide concentrations in wastewater as high as 200 mg/litre (Siller & Winter, 1998).
The major sources of cyanide released to air, in addition to exhaust from vehicle emissions, are diverse, including chemical manufacturing (hydrogen cyanide, methyl methacrylate, acrylonitrile); processing industries, such as metallurgical industries and metal plating (i.e., electroplating metals and finishing [metal polishes]); extraction of gold and silver from low-grade ores; volatilization from cyanide wastes disposed of in landfills and waste ponds; the production of coke or other coal carbonization procedures; emissions from municipal solid waste incinerators; and direct release of cyanides to the atmosphere resulting from fumigation operations, combustion of polyurethanes, acrylonitrile, and polyamide plastics, and combustion of wool, silk, and fibres (Carotti & Kaiser, 1972; Fiksel et al., 1981; ATSDR, 1997; Eisler et al., 1999).
An estimated total of 1 million tonnes of hydrogen cyanide, amounting to 73.1% of the total environmental releases in the USA, was discharged to the air from manufacturing and processing facilities (ATSDR, 1997).
The estimated amounts of hydrogen cyanide released to air in 1976 from the most common non-industrial sources were as follows: agricultural pest control, 62 tonnes; incineration, 8.2–82 tonnes; and tobacco smoke, 5.9–340 tonnes (Fiksel et al., 1981; ATSDR, 1997).
In 2001, from various locations in the USA, about 1300 tonnes of hydrogen cyanide were released on- and off-site; 540 tonnes were emitted to the atmosphere, 0.1 tonne was released to surface waters, 770 tonnes were injected into Class I wells,3 and 0.42 tonne was released to land (US EPA, 2003c). In 2001, from various locations in the USA, approximately 3400 tonnes of cyanides (not otherwise specified) were released on- and off-site; 220 tonnes were emitted to the atmosphere, 47 tonnes were released to surface waters, 1800 tonnes were injected into Class I wells, and 1300 tonnes were released to land (US EPA, 2003c).
Hydrogen cyanide has been found following the combustion of a number of synthetic polymers. The maximum yield of hydrogen cyanide per gram of polyurethane foam ranged from 0.37 to 0.93 mg under non-flaming conditions and from 0.5 to 1.02 mg under flaming combustion (Sklarew & Hayes, 1984). Hydrogen cyanide concentrations in the off-gas from the shale oil retorting process ranged from 7 to 44 mg/m3 (Sklarew & Hayes, 1984).
One cigarette without a filter liberates 500 µg hydrogen cyanide, while filter cigarettes liberate only 100 µg in mainstream smoke. Hydrogen cyanide concentrations in mainstream and sidestream smoke ranging from 280 to 550 µg/cigarette and from 53 to 111 µg/cigarette, respectively, have been reported; sidestream:mainstream ratios of hydrogen cyanide concentrations ranged from 0.06 to 0.50 (ATSDR, 1997). The level of hydrogen cyanide found in Canadian cigarette smoke under International Organization for Standardization standard smoking conditions were as follows: mainstream smoke, 32–156 µg/cigarette; and sidestream smoke, 77–136 µg/cigarette (Health Canada, 2002).
The average rate of emission of hydrogen cyanide by automobile exhaust was reported to be 7–9 mg/km for cars not equipped with catalytic converters and on the order of 0.6 mg/km for cars with catalytic converters operating under optimum conditions in the mid- to late 1970s (ATSDR, 1997).
Cyanogen chloride is formed as a reaction product of organic precursors with hypochlorous acid in the presence of ammonia and may be formed as a by-product of the chloramination of water (e.g., via the reaction of humic substances with chlorine and chloramine used for water disinfection) (Ohya & Kanno 1987; WHO, 1996; IPCS, 2000a). In the USA, 35% of the surface water plants and 23% of the groundwater plants using chloramine as a primary or secondary disinfectant report cyanogen chloride formation (US EPA, 2002).
Cyanogen is generated in the combustion of nitrogen–carbon compounds and appears in automobile exhaust gases and gases from blast furnaces (CHEMINFO, 1998).
Cyanide is present in the air mostly as a gas, and cyanides have the potential to be transported over long distances from their respective emission sources.
Cyanide is found in ambient air as hydrogen cyanide and to a smaller extent in particulate matter. The concentration of hydrogen cyanide measured since 1981 in the northern hemisphere’s non-urban troposphere ranged from 180 to 190 ng/m3 (Cicerone & Zellner, 1983; Jaramillo et al., 1989).
Ambient air monitoring data for cyanides in Bulgaria in areas near petrochemical plants showed concentrations ranging from 0.2 to 0.8 µg/m3 (annual average value) (Kaloyanova et al., 1985).
Cyanide has been detected at levels of 20–46 mg/m3 in the air near large-scale cassava processing facilities in Nigeria (Okafor & Maduagwu, 2000).
Cyanides, reported as cyanide, hydrogen cyanide, sodium cyanide, potassium cyanide, calcium cyanide, or copper(I) cyanide, have been detected in surface water samples at 70 of the 154 hazardous waste sites where they were studied in the USA; they have also been detected in groundwater samples at 191 of the 419 waste sites studied and in leachate samples of 16 of the 52 sites studied. The median concentrations in the positive samples were 160 µg/litre for groundwater, 70 µg/litre for surface water, and 479 µg/litre for the leachates (HazDat, 2003).
Data from the US National Urban Runoff Program in 1982 revealed that 16% of urban runoff samples collected from four cities across the USA contained cyanides at levels of 2–33 µg/litre (ATSDR, 1997).
According to the US Environmental Protection Agency’s (EPA) STORET database, the mean cyanide concentration in most surface waters in the USA is less than 3.5 µg/litre. Data from the late 1970s to early 1980s indicated that the levels are higher only in limited areas and may exceed 200 µg/litre (ATSDR, 1997).
In 1978, a US EPA survey of drinking-water supplies showed that about 7% of the supplies had cyanide concentrations greater than 10 µg/litre (US EPA, 1993a). Cyanogen chloride is one of the 18 compounds that occur most frequently (8 of 10 city surveys) in potable water within the framework of the US National Organic Reconnaissance Survey (Bedding et al., 1982). In a survey in 1987 of over 35 drinking-water supplies, the quarterly median cyanogen chloride concentrations in drinking-water ranged from 0.45 to 0.80 µg/litre (from 0.19 to 0.34 µg cyanide/litre) (Krasner et al., 1989; ATSDR, 1997). More current data regarding the cyanide and cyanogen chloride levels in drinking-water are lacking.
Levels of 1.58–7.89 mg cyanide/litre have been found in natural water sources near large-scale cassava processing facilities in Nigeria (Okafor et al., 2001).
Cyanide has been identified in the soil of hazardous waste sites in the USA; the median concentrations for the positive sites were 0.8 mg/kg in the subsurface soil (found at 77 sites of the 124 studied) and 0.4 mg/kg in the topsoil (51 positive sites out of 91 sites) (HazDat, 2003).
Cyanide-containing wastes are commonly found in soils at former manufactured gas plant sites in the USA. Most concentrations of cyanide compounds at the manufactured gas plant sites are below 2000 mg/kg. The most prevalent types of cyanide compounds are iron-complexed forms, e.g., ferric ferrocyanide (Prussian blue), rather than the highly toxic free cyanide forms. Iron-complexed cyanides, dominated by the ferrocyanide ion, comprise over 97% of total cyanides in either weathered or unweathered soils (Shifrin et al., 1996).
Many edible plants contain cyanogenic glycosides, whose concentrations can vary widely as a result of genetic and environmental factors, location, season, and soil types (Ermans et al., 1980; JECFA, 1993). Some of the foodstuffs and their cyanide contents are shown in Table 3. Cassava tubers vary widely in their cyanogenic glycoside content, although most varieties contain 15–400 mg cyanide/kg fresh weight. Occasionally varieties of cassava tubers contain 1300–2000 mg cyanide/kg fresh weight, and cassava leaves contain 1000–2000 mg cyanogenic glucosides/kg on a dry matter basis (Padmaja, 1995). Fermentation of cassava pulp for 96 h during gari production reduced the hydrogen cyanide content by 50%; soaking of sliced cassava for 24 h, 40%; and sun-drying, some 15% (Kendirim et al., 1995). It should be noted that the ranges of cyanide concentrations shown in Table 3 are very broad in several cases (i.e., cereals and their products, soy protein products, and apricot pits), which may be due to their different sources and differences in analytical procedures; as well, the values may reflect the older literature.
Hydrogen cyanide can be produced by hydrolytic reaction catalysed by one or more enzymes from the plants containing cyanogenic glycosides. In kernels, for example, this reaction is catalysed by the enzyme emulsin (Lasch & El Shawa, 1981) when the seeds are crushed and moistened. Amygdalin (which is also present in cassava, bitter almonds, and peach stones) is converted to glucose, benzaldehyde, and hydrogen cyanide (Figure 2) (IPCS, 1992). Hydrogen cyanide release can occur during maceration, which activates intracellular beta-glucosidases. This reaction can also result from chewing, which causes the enzyme and the cyanogenic glycosides stored in different compartments to combine (Ermans et al., 1980; Nahrstedt, 1993). The reaction occurs rapidly in an alkaline environment, and the hydrolysis is complete in 10 min. Hydrolysis is possible in an acid solution and takes place slowly.
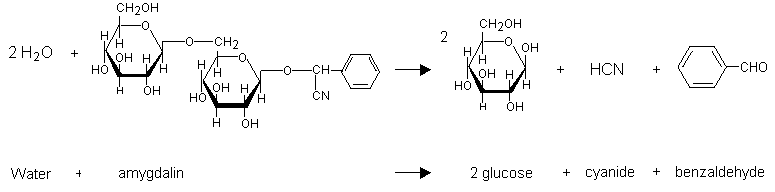
Fig. 2: Hydrolysis of amygdalin
Liberation of hydrogen cyanide from cyanogenic glycosides occurs usually after ingestion and hydrolysis by the glycosidases of the intestinal microflora and, to a lesser degree, by glucosidases of the liver and other tissues (Padmaja, 1995). However, hydrolysis may also occur during the preparation of the food, which may account for the short interval between ingestion and the appearance of signs of poisoning in some accidents (Lasch & El Shawa, 1981).
Table 3: Cyanide concentrations in food products.a
| Type of product | Cyanide concentration (in mg/kg or mg/litre) |
| Cereal grains and their products | 0.001–0.45 |
| Soy protein products | 0.07–0.3 |
| Soybean hulls | 1.24 |
| Apricot pits, wet weight | 89–2170 |
| Home-made cherry juice from pitted fruits | 5.1 |
| Home-made cherry juice containing 100% crushed pits | 23 |
| Commercial fruit juices | |
| Cherry | 4.6 |
| Apricot | 2.2 |
| Prune | 1.9 |
| Tropical foodstuffs | |
| Cassava (bitter) / dried root cortex | 2360 |
| Cassava (bitter) / leaves | 300 |
| Cassava (bitter) / whole tubers | 380 |
| Cassava (sweet) / leaves | 451 |
| Cassava (sweet) / whole tubers | 445 |
| Gari flour (Nigeria) | 10.6–22.1 |
| Sorghum / whole immature plant | 2400 |
| Bamboo / immature shoot tip | 7700 |
| Lima beans from Java (coloured) | 3000 |
| Lima beans fom Puerto Rico (black) | 2900 |
| Lima beans from Burma (white) | 2000 |
a From Nartey (1980); Honig et al. (1983); JECFA (1993); ATSDR (1997).
Hydrogen cyanide (HCN) is a colorless, rapidly acting, highly poisonous gas or liquid that has an odor of bitter almonds. Most HCN is used as an intermediate at the site of production. Major uses include the manufacture of nylons, plastics, and fumigants. Exposures to HCN may occur in industrial situations as well as from cigarette smoke, combustion products, and naturally occurring cyanide compounds in foods. Sodium nitroprusside (Na2[Fe(CN)5 NO]·2H2O), which has been used as an antihypertensive in humans, breaks down into nonionized HCN.
HCN is a systemic poison; toxicity is due to inhibition of cytochrome oxidase, which prevents cellular utilization of oxygen. Inhibition of the terminal step of electron transport in cells of the brain results in loss of consciousness, respiratory arrest, and ultimately, death. Stimulation of the chemoreceptors of the carotid and aortic bodies produces a brief period of hyperpnea; cardiac irregularities may also occur. The biochemical mechanisms of cyanide action are the same for all mammalian species. HCN is metabolized by the enzyme rhodanese which catalyzes the transfer of sulfur from thiosulfate to cyanide to yield the relatively nontoxic thiocyanate.
Human exposures with measured concentrations were limited to occupational reports. Symptoms of exposed workers ranged from no adverse health effects to mild discomfort to frank central nervous system effects. Repeated or chronic exposures have resulted in hypothyroidism. Inhalation studies resulting in sublethal effects, such as incapacitation, and changes in respiratory and cardiac parameters were described for the monkey, dog, rat, and mouse; lethality studies were available for the rat, mouse, and rabbit. Exposure durations ranged from a few seconds to 24 hours (h). Regression analyses of the exposure duration-concentration relationships for both incapacitation and lethality for the monkey determined that the relationship is C2×t= k and that the relationship for lethality based on rat data is C2.6×t=k.
The AEGL-1 is based on human monitoring studies in which the preponderance of data as a weight-of-evidence consideration indicates that an 8-h exposure to HCN at 1 parts per million (ppm) would be without adverse health effects for the general population. Although the exposures were of chronic duration (generally 8 h/day (d) for extended work periods) and the data are lacking in various aspects of specific exposure concentrations and well-documented exposure-related symptoms, it is human data which are most relevant in determining the AEGL-1 threshold of notable discomfort.
Chronic exposures (5–15 years [y]) in three electroplating plants to mean concentrations of 6, 8, and 10 ppm produced exposure-related symptoms including headache, weakness, and objectionable changes in taste and smell (El Ghawabi et al. 1975), but the authors failed to relate symptoms to air concentrations. Over half of the workers presented with enlarged thyroids (characteristically observed in cases of chronic cyanide exposure), which may have been responsible for certain symptoms. In evaluating the El Ghawabi et al. (1975) study, a National Research Council (NRC) subcommittee concluded that a 1-h exposure at 8 ppm would cause no more than mild headache in healthy adults (NRC 2000). Mild headache meets the definition of the AEGL-1. Chronic exposures of 63 healthy adult cyanide-production workers to geometric mean concentrations of ≤1 ppm of HCN (range, 0.01–3.3 ppm; measured with personal samplers), with potential exposures at 6 ppm (as measured with area samples), for part of a year resulted in no exposure-related adverse health effects (Leeser et al. 1990). Finally, although health effects were not specifically addressed, workers in five apricot kernel processing plants were exposed to air concentrations of HCN at <1 to 17 ppm (Grabois 1954). The fact that engineering controls were recommended “where required” at a time when the maximum allowable concentration was 10 ppm suggests that no untoward effects were occurring at the lower concentrations. The National Institute for Occupational Safety and Health (NIOSH) concluded from the Grabois (1954) data that 5 ppm was a no-effect concentration in an occupational setting (NIOSH 1976). Additional monitoring studies indicated that workers were routinely exposed to HCN at 4 to 6 ppm (Hardy et al. 1950; Maehly and Swensson 1970). Humans may differ in their sensitivity to the effects of HCN, but no data regarding specific differences among individuals were located in the available literature (occupational monitoring studies and the clinical use of nitroprusside solutions to treat chronic hypertension). The detoxifying enzyme rhodanese is present in large amounts in all individuals, including newborns. Because no specific susceptible populations were described following chronic exposures or during use of nitroprusside solutions to treat chronic hypertension, the potential differences in susceptibility among humans are not expected to exceed 3-fold.
The 8-h AEGL-1 value was derived from a consideration of the dose-response data obtained from all of the monitoring studies cited and subsequently time-scaled to the shorter AEGL exposure durations. Although the exposures were of chronic duration in all studies, they represent the only viable human data available. Furthermore, because symptoms observed or reported at given concentrations for the multiple 8-h exposures of a typical work schedule should represent the greatest potential responses, the use of the data represents a conservative approach to AEGL derivation. All of the exposure durations reported exceed the AEGL exposure durations, so the longest, or 8-h, AEGL exposure duration was selected as the basis for AEGL development. Dividing the 8-h concentration of 5 ppm from the Grabois (1954), Hardy et al. (1950), or Maehly and Swensson (1970) studies by an intraspecies uncertainty factor (UF) of 3 or dividing the 1-h concentration of 8 ppm from the El Ghawabi et al. (1975) study by an intraspecies UF of 3 result in very similar AEGL-1 values. The resulting 8-h value of 1.7 ppm is also similar to the 8-h geometric mean value of 1 ppm in the Leeser et al. (1990) study that was derived without application of a UF. A UF should not be applied to the Leeser et al. (1990) study, because it was the lowest no-observed-adverseeffect level (NOAEL). Using the 8-h value of 1 ppm as the basis for time scaling to shorter durations, the conservative relationship of C3×t=k was chosen for the derivations. The 10-minute (min) AEGL-1 was set equal to the 30-min value so as not to exceed the highest personal exposure concentration of 3.3 ppm in the well-conducted Leeser et al. (1990) study.
The AEGL-2 was based on an exposure of cynomolgus monkeys to a concentration of HCN at 60 ppm for 30 min, which resulted in a slight increase in the respiratory minute volume near the end of the exposure and a slight depressive effect on the central nervous system as evidenced by changes in electroencephalograms, also near the end of the exposure; there was no physiological response (Purser 1984). The mechanism of action of HCN is the same for all mammalian species, but the rapidity and intensity of the toxic effect is related to relative respiration rates as well as pharmacokinetic considerations. Based on relative respiration rates, the uptake of HCN by the monkey is more rapid than that of humans. The monkey is an appropriate model for extrapolation to humans because, compared with rodents, the respiratory systems of monkeys and humans are more similar in gross anatomy, the amount and distribution of types of respiratory epithelium, and airflow pattern. Because the monkey is an appropriate model for humans but is potentially more susceptible to the action of cyanide based on relative respiration rates, an interspecies UF of 2 was applied. Humans may differ in their sensitivity to HCN, but no data regarding specific differences among humans were located in the available literature. The detoxifying enzyme rhodanese is present in all individuals, including newborns. Therefore, an intraspecies UF of 3 was applied. The 30-min concentration of 60 ppm from the Purser (1984) study was divided by a combined interspecies and intraspecies UF of 6 and scaled across time for the AEGL-specified exposure periods using the relationship C2×t=k. The safety of the 30-min and 1-h values of 10 and 7.1 ppm, respectively, is supported by monitoring studies in which chronic exposures to average concentrations of 8 to 10 ppm may have produced primarily reversible central nervous system effects such as headaches in some workers (El Ghawabi et al. 1975).
The rat provided the only data set for calculation of LC01 values for different time periods (E.I. du Pont de Nemours and Company 1981). The LC01 values were considered the threshold for lethality and were used as the basis for deriving AEGL-3 values. The mouse, rat, and rabbit were equally sensitive to the lethal effects of HCN, as determined by similar LC50 values for the same time periods (for example, 30-min LC50 values of 166, 177, and 189 ppm for the mouse, rat, and rabbit, respectively). In an earlier study, times to death for several animal species showed that mice and rats may be slightly more sensitive to HCN than monkeys (and presumably humans). The differences in sensitivity were attributed, at least partially, to the more rapid respiratory rate of the rodent compared to body weight. Because LC50 values for several species were within a factor of 1.5 of each other and the respiration rate of rodents is higher than that of humans, resulting in more rapid uptake of HCN, an interspecies UF of 2 was applied. Humans may differ in their sensitivity to HCN, but no data regarding specific differences among humans were located in the available literature. The detoxifying enzyme rhodanese is present in all individuals, including newborns. Therefore, an intraspecies UF of 3 was applied to protect sensitive individuals. The 15- and 30-min and 1-h LC01 values (138, 127 and 88 ppm, respectively) were divided by a total UF of 6. The 15-min LC01 value was time scaled to 10 min to derive the 10-min AEGL-3; the 30-min LC01 was used for the 30-min AEGL-3 value; and the 60-min LC01 was used to calculate the 1-, 4-, and 8-h AEGL-3 concentrations. For the AEGL-3 values, scaling across time utilized empirical data (i.e., the lethal concentration-exposure duration relationship for the rat in the key study, C2.6 ×t=k). The safety of the 4- and 8-h AEGL-3 values of 8.6 and 6.6 ppm is supported by the lack of severe adverse effects in healthy workers chronically exposed to similar values during monitoring studies (Grabois 1954; El Ghawabi et al. 1975). The values appear in Table 5–1.
TABLE 5–1
Summary Table of AEGL Values for Hydrogen Cyanide (ppm [mg/m3]).
I. INTRODUCTION
Hydrogen cyanide (HCN) is a colorless, highly poisonous gas or liquid (below 26.7 °C) having an odor of bitter almonds (Hartung 1994; Pesce 1994). It is a weak acid. Exposures may occur in industrial situations as well as from cigarette smoke and combustion products and from naturally occurring cyanide compounds in foods. There is a potential for exposure when any acid is mixed with a cyanide salt. Intravenously administered sodium nitroprusside (Na2[Fe(CN)5NO]·2H2O) has been used clinically to lower blood pressure (Schulz et al. 1982). Chemical and physical properties are listed in Table 5–2.
TABLE 5–2
Chemical and Physical Data.
HCN is produced commercially by the reaction of ammonia, methane, and air over a platinum catalyst or from the reaction of ammonia and methane. HCN is also obtained as a by-product in the manufacture of acrylonitrile and may be generated during many other manufacturing processes (Pesce 1994). In 1999, there were 34 companies operating 47 HCN production facilities in the United States, Western Europe, and Japan (CEH 2000). The estimated production capacity was 3.5 billion pounds. The demand for HCN is expected to increase by 2.8% per year through 2004.
Most HCN is used at the production site (CEH 2000). HCN is widely used; according to Hartung (1994), the major uses are in the fumigation of ships, buildings, orchards, and various foods; the production of various resin monomers such as acrylates, methacrylates, and hexamethylenediamine; and the production of nitriles. HCN may also be generated during the use of cyanide salts in electroplating operations and mining. Pesce (1994) estimated the following usage percentages: adiponitrile for nylon, 41%; acetone cyanohydrin for acrylic plastics, 28%; sodium cyanide for gold recovery, 13%; cyanuric chloride for pesticides and other agricultural products, 9%; chelating agents such as EDTA, 4%; and methionine for animal feed, 2%. CEH (2000) lists the following three dominant products: acetone cyanohydrin (for methyl methacrylate), adiponitrile (for hexamethylenediamine), and sodium cyanide (used as a reagent).
The U.S. Department of Transportation subjects HCN to rigid packaging, labeling, and shipping regulations. HCN can be purchased in cylinders ranging from 300 mL to 75 kg. Tank car sizes are 24 and 46 tons. Since 1950, there have been no accidents during the bulk transportation of HCN (Pesce 1994). HCN is usually shipped as a water solution containing a stabilizer of 0.05% phosphoric acid (HSDB 2000).
2. HUMAN TOXICITY DATA
HCN is among the most rapidly acting of all known poisons. Absorption occurs by all routes; the mechanism of action is inhibition of cellular respiration. The respiratory, central nervous, and cardiovascular systems are the primary targets of an acute exposure. Information on human exposures was limited to exposures to high concentrations for short time intervals, poorly documented accidental exposures, and chronic occupational exposures.
According to Hartung (1994), a few breaths at “high concentrations” may be followed by rapid collapse and cessation of respiration. If the exposure continues, unconsciousness is followed by death. At much lower concentrations, the earliest symptoms may be numbness, weakness, vertigo, some nausea, and rapid pulse. The respiratory rate increases initially and at later stages becomes slow and gasping. Chronic exposures have been related to thyroid enlargement. Cardiac effects include electrocardiogram changes (HSDB 2000). HCN is not considered a lacrimator (Weedon et al. 1940). Should individuals survive the acute phase of HCN intoxication, recovery can be uneventful and without permanent sequelae.
In addition to occupational exposures, humans are exposed to cyanide in their diets (from cyanide- and amygdalin-containing foods and fumigation residues) and through cigarette smoke, automobile exhaust, and fires (NIOSH 1976; HSDB 2000). Exposure from smoking is not trivial; each puff from an unfiltered cigarette, which contains 35 μg of HCN, momentarily exposes the lung to a concentration of approximately 46 ppm (Carson et al. 1981). Yamanaka et al. (1991) reported that mainstream cigarette smoke contains HCN at 40–70 ppm, and side-stream smoke contains less than 5 ppm.
The odor of HCN has been described as that of bitter almond. The ability to detect the odor varies widely and about 20% of the population is genetically unable to discern this characteristic odor (Snodgrass 1996). A review of literature on odor thresholds revealed that the odor threshold for HCN can range from 0.58 to 5 ppm (Amoore and Hautala 1983; Ruth 1986). An irritating concentration was not reported.
2.1. Acute Lethality
Although a great many deaths have occurred from accidental, intentional, or occupational exposures to HCN, in only a few cases are specific exposure concentrations known. In a review of human fatalities (ATSDR 1997), it was stated that exposure to airborne concentrations of HCN at 180 to 270 ppm were fatal, usually within several minutes, and a concentration of 135 ppm was fatal after 30 min. The average fatal concentration for humans was estimated at 546 ppm for 10 min. The latter data point is based on the work of McNamara (1976), who considered the resistance of man to HCN to be similar to that of the goat and monkey and four times that of the mouse. Fatal levels of HCN cause a brief period of central nervous system stimulation followed by depression, convulsions, coma with abolished deep reflexes and dilated pupils, and death. Several review sources, such as Dudley et al. (1942), Hartung (1994), and ATSDR (1997), report human toxicity data that appear to be based largely on pre-1920 animal data.
2.2. Nonlethal Toxicity
Several studies of occupational exposures and one study with a human subject were located. In the occupational exposures (summarized in Table 5– 3), neurological symptoms consistent with cyanide intoxication were demonstrated, but the likelihood of concomitant exposure to other chemicals could not be ruled out. For example, cleaners and cutting oils, as well as sodium and copper cyanide, may be present in electroplating operations (ATSDR 1997). The experimental human study involved the exposure of a single subject and a dog to a high concentration for a short exposure period.
TABLE 5–3
Occupational Exposures to Hydrogen Cyanide.
Adverse health consequences on systems other than the central nervous and respiratory systems have been documented during occupational and/or accidental exposures. Generally, these effects occurred following chronic exposures, but the cardiovascular and dermal effects could occur following acute exposures. For example, cardiovascular effects (palpitations, hypotension, and chest pain) (El Ghawabi et al. 1975; Blanc et al. 1985; Peden et al. 1986), hematological effects (increased or decreased hemoglobin) (El Ghawabi et al. 1975; Kumar et al. 1992), hepatic effects (increased serum alkaline phosphatase activity but not serum bilirubin) (Kumar et al. 1992), gastrointestinal effects (nausea and vomiting) (El Ghawabi et al. 1975), endocrine effects (thyroid enlargement) (Hardy et al. 1950; El Ghawabi et al. 1975; Blanc et al. 1985), and dermal effects (burns and rashes) (Blanc et al. 1985; Singh et al. 1989) have been observed. Authors of several studies, including Hardy et al. (1950), observed that some of the symptoms of chronic cyanide exposure are a result of thiocyanate-induced goiter. These authors noted that goiter has also been reported following thiocyanate therapy for hypertension.
El Ghawabi et al. (1975) compared the symptoms of 36 workers exposed to HCN in three electroplating factories in Egypt with a referent group; employment ranged between 5 and 15 y. None of the workers in either the exposed or control groups were smokers. Cyanide exposure resulted from a plating bath that contained copper cyanide, sodium cyanide, and sodium carbonate. Concentrations of cyanide in the breathing zone of the workers ranged from 4.2 to 12.4 ppm (means in the three factories: 6, 8, and 10 ppm). Fifteen-minute air samples were collected in NaOH and analyzed colorimetrically. Symptoms reported most frequently by exposed workers compared with the referent control group were, in descending order of frequency: headache, weakness, and changes in taste and smell. Lachrimation, vomiting, abdominal colic, precordial pain, salivation, and nervous instability were less common. The authors made no attempt to correlate the incidences of these symptoms with concentrations. Although there were no clinical manifestations of hypoor hyperthyroidism, 20 of the workers had thyroid enlargement to a mild or moderate degree; this conditions was accompanied by higher 131I uptake compared with the referent controls. Exposed workers also had significantly higher blood hemoglobin, lymphocyte cell counts, cyanmethemhemoglobin, and urinary thiocyanate levels than controls. Urinary thiocyanate levels were correlated with cyanide concentration in workplace air. Two workers in the factory with a mean exposure of 10 ppm suffered psychotic episodes; recovery occurred within 36 to 48 h. Although the sample size was small, the study used well-matched controls and included a biological index of exposure (urinary thiocyanate). The NRC Subcommittee on Spacecraft Maximum Allowable Concentrations, in evaluating the El Ghawabi et al. (1975) data, concluded that “8 ppm would likely produce no more than mild CNS effects (e.g., mild headache) which would be acceptable for 1-hour exposures” of healthy adults (NRC 2000). ATSDR (1997) noted that exposure to cleaners and cutting oils may have contributed to the effects observed in this study.
Grabois (1954) surveyed HCN levels in five plants that processed apricot kernels in order to determine possible health hazards. The survey was performed by the Division of Industrial Hygiene of the New York State Department of Labor. Work area concentrations in the plants ranged from <1 to 17 ppm, and two areas in one of the plants had levels of 17.0 ppm (comminuting area) and 13.9 ppm (cooking area). The general workroom atmosphere in this plant averaged a 6.4 ppm concentration of HCN. Medical questionnaires were not given and the health status of the employees was not reported. However, recommendations were made for controlling HCN exposures “where required,” presumably where concentrations were above the then maximum recommended concentration of 10 ppm. NIOSH (1976), in interpreting the Grabois (1954) data, stated that 5 ppm was a no-effect level, and higher concentrations were only rarely present.
Chandra et al. (1980) studied the effects of HCN exposure on 23 male workers engaged in electroplating and case hardening. The workers avoided cyanogenic foods such as cabbage and almonds for 48 h prior to blood and urine sampling. In spite of the low exposure levels—0.8 mg/m3 (0.7 ppm) in the breathing zone and 0.2 mg/m3 (0.2 ppm) in the general work area—the workers complained of typical symptoms of HCN poisoning (symptoms not stated); however, no objective measures of adverse health effects were reported. Higher blood and urine cyanide and thiocyanate were measured in exposed workers compared with a control group. Higher levels of blood and urine cyanide and thiocyanate were present in smokers than in nonsmokers in both the exposed and control groups.
Blanc et al. (1985) surveyed and examined 36 former employees of a silver reclaiming facility in order to determine acute and potential residual adverse health effects resulting from occupational HCN exposure. The study was prompted by a worker fatality from acute cyanide poisoning. The workers had been exposed long-term to excessive concentrations of cyanide as the time-weighted average (TWA) taken 24 h after the plant had closed down was 15 ppm. The most frequently reported symptoms included headache, dizziness, nausea or vomiting, almond or bitter taste, eye irritation, loss of appetite, epistaxis, fatigue, and rash. The most prevalent symptoms (headache, dizziness, nausea or vomiting, and a bitter or almond taste) were consistent with acute cyanide poisoning. A concentration-response relationship corresponding to high- and low-exposure jobs was demonstrated, but exact breathing zone concentrations were unknown. Some symptoms exhibiting a dose-response trend occurring seven or more months after exposure had ceased. Mild abnormalities of vitamin B12, folate, and thyroid function were detected and suggested long-term cyanide and thiocyanate involvement. The NRC (2000), in reviewing this study, pointed out that the 24-h TWA of 15 ppm was measured one day after the plant had closed down, suggesting that workers may have been exposed to cyanide at more than 15 ppm.
Hardy et al. (1950) observed increased urinary excretion of thiocyanate in a group of case-hardener workers (hot metals are dropped into baths of cyanide salts in order to harden the material). Two workers with unqualified exposures suffered persistent headaches, sweating, chest pains, dizziness, fatigue, weakness, mental confusion, disturbed motor function, nervousness, coughing, sneezing, cramping in the lower abdomen, auricular fibrillation, and thyroid enlargement. The authors indicated that ≤10 ppm should prevent cyanide toxicity in workers, and with adequate engineering controls, workers were routinely exposed at 4–6 ppm. No symptoms were surveyed or discussed for these routine exposures.
Radojicic (1973) reported fatigue, headache, weakness, tremor in the arms and legs, pain, and nausea in 28 electroplating workers and 15 foundry workers chronically exposed to cyanide. Employment duration ranged from 0 to 15 y. Area atmospheric concentrations ranged from 6 to 13 ppm in the electroplating facility (four measurements) and 5 to 8 ppm in the foundry (three measurements). In the electroplating facility, higher concentrations were measured over work vats, 10 to 13 ppm, than in the middle of the room where concentrations were 6 to 8 ppm. In both facilities, urinary thiocyanate levels of workers were higher after work than prior to work, were higher in smokers than in nonsmokers, and increased with the number of years of work. Urinary thiocyanate concentrations were higher in smokers prior to a work day than in nonsmokers following a work shift. Symptoms were more pronounced in workers with the longer exposures.
Urinary and blood cyanide and thiocyanate were measured in a group of 140 workers consisting of exposed and nonexposed smokers and nonsmokers (Maehly and Swensson 1970). The HCN-exposed group consisted of 39 nonsmokers and 55 smokers. Area measurements, sampled with Draeger tubes at each work station, ranged from 1 to 10 ppm (average, 5 ppm). Blood and urinary cyanide and thiocyanate levels varied widely among the groups, and there was no clear relationship to occupational exposure at these concentrations; blood cyanide levels did not bear a relationship to exposure via smoking, but free thiocyanate levels in the urine tended to be higher in smokers than in nonsmokers. No worker symptoms were reported in this study.
Leeser et al. (1990) reported a cross-sectional study of the health of cyanide-salt production workers. Sixty-three cyanide production workers employed for 1 to 40 y were compared with 100 referent workers from a diphenyl oxide plant. Workers were examined before and after a block of six 8-h shifts. All workers had full medical examinations, routine clinical chemistry tests, and blood samples taken for measurement of blood cyanide and carboxyhemoglobin. In addition, circulating levels of vitamin B12 and thyroxine (T4) were measured. Atmospheric cyanide was monitored with static monitors, Draeger pump tests, and personal monitoring. For the personal monitoring, air was drawn through bubblers which contained sodium hydroxide. Cyanide collected in the sodium hydroxide solution was measured using an anion-selective ion electrode. All results (a total of 34 samples) were between 0.01 and 3.6 mg/m3 (0.01 and 3.3 ppm). Geometric mean values for eight job categories ranged between 0.03 and 1.05 mg/m3 (0.03 and 0.96 ppm). Values for only one job category (eight personal samples) averaged 0.96 ppm. Results of routine Draeger pump tests (area samples) were between 1 and 3 ppm (none were above 10 ppm). In addition, during the fall of the year, production problems in part of the plant caused the HCN level to increase to 6 ppm from the usual 1–3 ppm (measurement method not stated). This increased exposure was reflected in an increase in mean blood cyanide level in the workers following a block of six 8-h shifts, and there was an increase of 5.83 μmol during the 6 ppm exposure compared with a decrease of 0.46 μmol across the shift block in the spring. Static monitors on all floors, set to trigger alarms at 10 ppm, did not sound during the study. Blood cyanide levels in exposed workers, though low, were generally higher than in control workers, and the highest levels were measured in cyanide-exposed nonsmokers compared with the nonsmoking control group (cyanide-exposed nonsmokers, 3.32 μmol; controls, 1.14 μmol; p<0.001). For ex-smokers, the difference was smaller (cyanide exposed, 2.16 μmol; controls, 1.46 μmol), and for current smokers, the blood cyanide level was actually higher in the control group (2.94 μmol for cyanide workers who smoked; 3.14 μmol for controls who smoked). The percentage of workers reporting symptoms such as shortness of breath and lack of energy was higher in cyanide workers than in the diphenyl oxide plant workers. These differences were partially explained by the greater number of cyanide workers who were shift workers. Slightly higher hemoglobin values and lymphocyte counts in the cyanide workers were not dose-related. Results of clinical and physical examinations and evaluation of medical histories failed to reveal any exposure-related health problems.
A 20-year-old man employed in a photographic darkroom suffered attacks of numbness, weakness, vertigo, some nausea, rapid pulse, and flushing of the face after 1 h of work (Parmenter 1926). Two other workers were unaffected. Following improved ventilation in the room, cyanide was measured in several areas of the workroom, including over a sink into which ferrous sulphate and potassium cyanide were routinely disposed. Concentrations of cyanide at that time (with the improved ventilation) ranged from 25 to 75 ppm.
During inspection of a tank containing a thin layer of hydrazodiisobutyronitrile (HZDN), a worker collapsed after 3 min, was fitted with a breathing apparatus after another 3 min, and removed from the tank after 13 min, resulting in a 6-min exposure (Bonsall 1984). At that time the worker was unconscious with imperceptible breathing and dilated pupils. He was covered with chemical residue. The tank had previously been washed with water; HZDN decomposes with water to give HCN and acetone. No HCN was measured prior to entry into the tank, but immediately after the incident, levels of HCN of about 500 mg/m3 (450 ppm) were measured. One hour after the exposure, the comatose individual was administered sodium thiosulfate, and following subsequent complications and treatment, he was discharged after 2 weeks (wk). No sequelae were apparent.
Barcroft (1931) described the controlled exposure of a 45-year-old, 70-kg man and a 12-kg dog to a concentration of HCN at 500–625 ppm in an airtight chamber. The human volunteer attempted to maintain the same level of activity as the dog. The dog became unsteady at 50 seconds (s), unconscious at 75 s, and convulsive at 90 s. One second later, the man walked out of the exposure chamber with no apparent effect. At 5 min after initiation of exposure, the man experienced a momentary feeling of nausea, and at 10 min from the start, his ability to concentrate in “close conversation” was altered. The dog at first appeared to be dead but recovered without adverse signs by the next day. Barcroft (1931) cites two other studies in which fumigation workers were exposed to a concentration of HCN at 250 ppm for 2 min or 350 ppm for 1.5 min without dizziness.
2.3. Developmental and Reproductive Effects
No data concerning developmental or reproductive effects of HCN in humans were identified in the available literature.
2.4. Genotoxicity
No data concerning the genotoxicity of HCN in humans were identified in the available literature.
2.5. Carcinogenicity
No data concerning the carcinogenicity of HCN in humans were identified in the available literature.
2.6. Summary
A great many human fatalities associated with acute HCN exposure have occurred, but exposure concentrations are for the most part unknown. Acute exposures that failed to result in mortality were either to high concentrations for very short exposure durations (approximately 500 or 450 ppm for approximately 1.5 min or 6 min, respectively [Barcroft 1931; Bonsall 1984]) or to exposure concentrations and times that were estimated (>25 ppm for about 1 h [Parmenter 1926]). Monitoring studies indicate that workers were routinely exposed at ≤10 ppm (Hardy et al. 1950; Grabois 1954; Maehly and Swensson 1970). Occupational HCN exposures at 1–10 ppm were acceptable at the time of these surveys as 10 ppm was the maximum acceptable concentration for workers. More effective exhaust ventilation was implemented “where required,” presumably where exposures were greater than 10 ppm, as in the Grabois (1954) study. The low exposures in the Leeser et al. (1990) study did not result in adverse health effects. Concentrations greater than 8–10 ppm may cause discomfort, and with long-term exposures, more serious symptoms can develop (El Ghawabi et al. 1975). The most common complaints in the monitoring study by El Ghawabi et al. (1975) were headache, weakness, and changes in taste and smell. Specific exposure levels for specific symptoms were not provided nor were concurrent exposures to other chemicals noted. Chronic exposure to low concentrations of HCN has been associated with hypothyroidism (development of goiter) (Hardy et al. 1950), and some symptoms associated with chronic exposures may be attributed to thyroid effects. It should be noted that in the study of Radojicic (1973) symptoms in workers increased with the number of years of work, and 20 of 36 workers in the study of El Ghawabi et al. (1975) had thyroid enlargement. No information on developmental and reproductive effects, genotoxicity, or carcinogenicity in humans was located.
3. ANIMAL TOXICITY DATA
NIOSH (1976) reviewed and summarized animal studies prior to 1976. Many of those studies are deficient in descriptions of exposure and analytical techniques as well as exposure concentrations and durations. Considerations of most of those pre-1976 studies are not reviewed here. Several of those earlier studies describe brain lesions in exposed animals. Histopathological examinations were performed in only a few of the studies conducted after 1976.
3.1. Acute Lethality
Acute inhalation lethality data for the rat, mouse, and rabbit for exposure times of 10 s to 12 h were located. A single inhalation study with the dog did not give an exposure duration. The data are summarized in Table 5–4. Data from studies with nonlethal concentrations are summarized in Table 5–5. Barcroft (1931) reported LC50 values and times to death for eight species of animals, the times to death at a constant concentration. Due to experimental design constraints, the LC50 values are not reported here, but relevant data are discussed in the section on relative species sensitivity (Section 4.4.1).
TABLE 5–4
Summary of Acute Lethal Inhalation Data in Laboratory Animals.
TABLE 5–5
Summary of Nonlethal Inhalation Data in Laboratory Animals.
3.1.1. Dogs
Dudley et al. (1942) cites a brief exposure to 115 ppm as fatal to dogs. Ninety parts per million may be tolerated for “hours” with death occurring after exposure. Exposures at 30–65 ppm for an unspecified duration led to vomiting, convulsions, and possibly death. No details on the source of the data, exposure durations, or experimental protocols were provided.
3.1.2. Rats
Groups of ten Wistar rats (gender not stated) inhaled concentrations of 280, 357, 368, 497, 583, or 690 ppm for 5 min in a Rochester chamber (Higgins et al. 1972). The animals were observed for 7 days (d) following exposure. A cage containing the animals was rapidly lowered into a chamber into which HCN was continuously delivered; the cage was rapidly removed after 5 min. HCN concentrations were continuously monitored using specific ion electrodes. All deaths occurred during the exposure period or within 20 min after exposure. The 5-min LC50 was 503 ppm (95% confidence limit (CL), 403–626 ppm). Using the same protocol, the 5-min LC50 for five male Sprague-Dawley rats was 484 ppm (95% CL, 442–535 ppm) (Vernot et al. 1977). Protocol details of the Vernot et al. (1977) study were not provided.
Groups of ten male Crl:CD rats were exposed to HCN in polymethymethacrylate exposure chambers under flow-through conditions (E.I. du Pont de Nemours 1981). The chamber atmosphere was measured continuously by infrared spectrophotometry; measurements were validated by gas chromatography. The experiment was performed in duplicate with one set of animals exposed head-only to the test gas while the other set was allowed free movement inside the exposure chamber. Free-moving rats inhaled concentrations of 273 to 508 ppm for 5 min, 110 to 403 ppm for 15 min, 128 to 306 ppm for 30 min, or 76 to 222 ppm for 60 min. The postexposure observation period was 14 d, during which body weights were monitored.
For all exposure durations, deaths occurred during exposures or within 1 d postexposure. The LC50 values for the 5-, 15-, 30-, and 60-min exposure periods for the unrestrained rats were 369 ppm (95% CL, 350–395 ppm), 196 ppm (95% CL, 181–209 ppm), 173 ppm (95% CL, 163–188 ppm), and 139 ppm (95% CL, 120–155 ppm), respectively. Using probit analysis, the authors also calculated LC01 values for the 5-, 15-, 30-, and 60-min exposure durations of 283, 138, 127, and 88 ppm, respectively. The LC50 values were lower (higher toxicity) for restrained rats: 398, 163, 85, and 63 ppm for the respective exposure durations.
Ballantyne (1983) exposed groups of six to ten rats to various concentrations of HCN for 10 s to 60 min. Lethal values are reported in Table 5–4; no further details of the study were reported. Kimmerle (1974), in citing his own unpublished data, reports 30- and 60-min LC50 values for the rat of 200 and 120 ppm, respectively. No details of the exposures were given.
Groups of six male Fischer 344 rats were exposed to various concentrations of HCN (not given) for 30 min (Levin et al. 1987). The rats were placed in restrainers for head-only exposures. Exposure chamber atmospheres were analyzed every 3 min with a gas chromatograph equipped with a thermionic detector. Most deaths occurred during the exposures. The 30-min LC50, calculated from deaths during the exposure period plus any deaths occurring up to 24 h postexposure, was 110 ppm with 95% CL of 97–127 ppm. It should be noted that LC50 values are lower for restrained animals than for unrestrained animals (E.I. du Pont de Nemours 1981).
Weedon et al. (1940) exposed groups of eight rats (strain not identified) to HCN at 1,000, 250, 63, or 16 ppm; times to 50% mortality (LT50) were recorded. Times to 50% mortality at the respective concentrations were as follows: 1.4, 8.7, 40, and >960 min.
Five male and five female Sprague-Dawley Crl:CD rats inhaled HCN at 68 ppm in a stainless steel chamber for 6 h/d for 3 d (Blank 1983). HCN was generated by passing nitrogen over the liquid contained in a 500-mL flask. The concentration in the cage was measured with an infrared analyzer. During the exposures, hypoactivity and rapid, shallow breathing were observed in all animals. During the first day, three males exhibited anoxia or hypoxia, followed by convulsions in one male. One male died during the exposure, a second male died during the postexposure observation period, and a third male was found dead prior to the second day of exposure. Two additional males and all five females exhibited breathing difficulties following the first exposure. No additional mortality was observed following the second and third days of exposure; body weights by the third day were below pre-exposure weights. Necropsy examinations of the three dead males revealed cyanosis of the extremities, moderate-to-severe hemorrhage of the lung, lung edema, tracheal edema, blanched appearance of the liver, singular occurrences of blood engorgement of the heart and surrounding vessels, chromorhinorrhea, urine-filled bladder, and gaseous distension of the gastrointestinal tract. Survivors were sacrificed following the last exposure. Of the seven survivors, three females exhibited slight-to-moderate hemorrhage of the lung.
3.1.3. Mice
Groups of 15 ICR mice (gender not stated) inhaled concentrations of 200, 283, 357, 368, 414, or 427 ppm for 5 min in a Rochester chamber (Higgins et al. 1972). The animals were observed for 7 d following exposure. Exposures were conducted in the same manner as for rats (Section 3.1.2). All deaths occurred during the exposure period or within 20 min after exposure. The 5-min LC50 was 323 ppm (95% CL, 276–377 ppm). The same data were reported in summary form by Vernot et al. (1977).
According to Matijak-Schaper and Alarie (1982), the 30-min LC50 of male Swiss-Webster mice inhaling HCN is 166 ppm. Mortality ratio for the mice (four per exposure group) were 0/4, 2/4, 3/4 and 4/4 for exposure to concentrations of HCN at 100, 150, 220, and 330 ppm, respectively. The recovery period was 10 min, during which the surviving mice appreciably recovered. The LC50 was the same for cannulated mice. At exposure concentrations of 500 and 750 ppm, the mean times to death were 12 min and 2 min, respectively.
Weedon et al. (1940) exposed groups of four mice (strain not identified) to HCN at 1,000, 250, 63, or 16 ppm and times to 50% mortality were recorded. Times to 50% mortality at the respective concentrations were: 1.2, 5.1, 66, and >960 min.
Groups often Swiss-Webster mice (both genders) inhaled HCN at concentrations of 30 ppm for 24 h, 100 ppm for 4 or 12 h, or 150 ppm for 4 h in flow-through chambers (Pryor et al. 1975). The temperature was 30°C and the atmosphere contained 21% oxygen. HCN was detected and quantified with detector tubes. All ten mice survived the 24 h exposure at 30 ppm; the postexposure period was 10 d. One mouse died during exposure at 100 ppm for 4 h, and all mice died from exposure at 100 ppm for 12 h and 150 ppm for 4 h. Although not specifically stated for HCN, it was indicated that all mice in the study, including those exposed to other gases, showed evidence of congestion of the lungs and vascular system. The authors noted the difficulty in attaining targeted concentrations of HCN in the chambers due to absorption on chamber surfaces; that difficulty was overcome by removing individual animal partitions from the exposure chamber.
3.1.4. Rabbits
Ballantyne (1983) exposed groups of six to ten rabbits to various concentrations of HCN for 45 s to 35 min. Values are reported in Table 5–4; no further details of the study were reported.
3.2. Nonlethal Toxicity
Toxicity studies resulting in nonlethal effects are reported in Table 5–5. Acute exposure data were available for the monkey, rat, and mouse with exposure durations ranging from 5 min to 24 h. Limited data were available for the dog.
3.2.1. Nonhuman Primates
Four cynomolgus monkeys (gender not stated) were individually exposed via a face mask to a concentration at 60 ppm for 30 min (Purser 1984). Each animal was exposed on three occasions. The same animals were used for hypoxia and hypercapnia tests. HCN, supplied from a standard gas mixture, was diluted with air; the concentration was measured intermittently using colorimetric tubes. Air flow into and out of the lungs was measured with a pneumotachograph connected to a differential gas pressure transducer. Several heart, blood, muscular, and central nervous system parameters were measured before, during, and after the exposures.
At 60 ppm, there was a slight depressive effect on the central nervous system, as evidenced by changes in brain wave activity at the end of the exposure periods (indicated in electroencephalograms [EEGs]), and the auditory cortical evoked potential (measured by electrodes on the surface of the auditory cortex) was reduced in amplitude during the late response. There was no physiological response to the EEG changes. There was a small increase in respiratory minute volume, but no adverse effects were observed on cardiovascular parameters or on neuromuscular conduction. The authors stated that concentrations of HCN below 60 ppm are unlikely to produce a significant impairment of escape capability.
In a follow-up study, four cynomolgus monkeys were individually exposed via a face mask to concentrations of HCN at 100 to 156 ppm for 30 min in order to measure time to incapacitation (“defined as a semiconscious state with loss of muscle tone”) (Purser et al. 1984). HCN was produced by introducing air, oxygen, and a mixture of HCN in nitrogen directly into the mixing chamber in proportions needed to produce the required atmospheric concentration; concentrations were estimated by silver nitrate titration from samples taken in 0.1 M sodium hydroxide solution. Several physiological parameters were measured before, during, and after the exposures. Results of earlier tests (not described) had determined at what concentration early signs of a physiological response occurred.
Time to incapacitation for the 100, 102, 123, 147, and 156 ppm concentrations were 19, 16, 15, 8, and 8 min, respectively; the relationship between exposure and time to incapacitation was linear. During exposures, effects consisted of hyperventilation (within 30 s), loss of consciousness, and bradycardia with arrhythmias and T-wave abnormalities; recoveries were rapid after exposure. The animal inhaling 147 ppm stopped breathing after 27 min and required resuscitation. Two additional exposures were terminated prior to the end of the 30 min due to severe signs. Animals rapidly recovered and were active during the first 10 min after exposure even though blood cyanide remained at levels that initially caused incapacitation. Purser (1984) states that the hyperventilatory response followed by incapacitation occurs at ≥80 ppm, but neither paper (Purser 1984; Purser et al. 1984) provides the experimental data for the 80 ppm concentration. At 180 ppm, hyperventilation occured almost immediately, and at 90 ppm the response was delayed for 20 min.
Although the primary mechanism of action of HCN is not respiratory irritation, the RD50—the concentration that produces a 50% decrease in respiratory rate—was measured in rats (E.I. du Pont de Nemours 1981). Respiratory rates were measured in restrained rats during all exposure durations (5–60 min). The RD50 was approximately 125 ppm. Although the RD50 may be considered in setting standards for primary irritants (to protect against sensory irritation), it is of limited use in setting standards for highly toxic, systemically acting chemicals. The highest concentrations that did not result in deaths of rats (see section 3.1.2 for details) are also listed in Table 5–5.
Six male Wistar rats inhaled HCN at 55 ppm for 30 min (Bhattacharya et al. 1984). HCN was generated by reaction of KCN with sulfuric acid and circulated through the chamber at the rate of 1 L/min. The rats were fitted with a lung mechanics analyzer (Buxco Electronic Inc.), and changes in air flow, transthoracic pressure, tidal volume, compliance, resistance, respiratory rate, and minute volume were determined every 10 min. Animals were sacrificed immediately following the exposure, and lungs were excised and analyzed for phospholipids (surfactant).
The authors stated that the exposure was “well tolerated” for the 30-min duration (Bhattacharya et al. 1984). With the exception of airway resistance, all lung dynamic parameters were significantly changed at 30 min, with increases in air flow, transthoracic pressure, and tidal volume and decreases in compliance, respiratory rate (60–70% decrease), and minute volume. There was a significant decrease in phospholipids in the lungs, but the toxicological relevance of that finding to AEGL derivation is not clear.
Five repeated exposures of 200 ppm for 12.5 min every 4 d resulted in increased cardiac-specific creatine phosphokinase activity in the blood (pooled data measured at 2 h after the first, third, and fifth exposures) and ectopic heart beats during the first 2 min after injection of norepinephrine (after the fifth exposure) but failed to induce cardiac lesions (histopathologic examinations at 14 d postexposure) (O'Flaherty and Thomas 1982). The rats were restrained and anesthetized.
Weedon et al. (1940) exposed groups of eight rats to a concentration of HCN at 16 ppm for 16 h. No deaths occurred, and rats appeared normal during the exposure. At autopsy of two rats, the lungs of one rat showed “pseudotuber-culosis.” All other organs in that rat and the other rat were normal.
3.2.4. Mice
Matijak-Schaper and Alarie (1982) measured the RD50 in four male Swiss-Webster mice. They pointed out that HCN is not primarily an irritant, and its mechanism of action is depression of the central respiratory center. The concentration that decreased the respiratory rate by 50% was 63 ppm (lower than the LC50 by a factor of 2.6). The exposure was for 30 min. Unconsciousness did not occur at this concentration (Alarie 1997). The RD50 for cannulated mice was 34 ppm, indicating that at least part of the respiratory decrease in noncannulated mice is due to sensory irritation. The breathing pattern of a mouse inhaling 80 ppm for 30 min was characterized as having “intermittent periods of sensory irritation…between segments of normal but slower breathing.” Time to asphyxia (as determined by respiratory pattern) at 150 ppm was 11 min. Times to asphyxia were not given for lower concentrations; however, “below the RD50 of 63 ppm, there were occasional breaths that were characteristic of asphyxiation, but this was a very transient occurrence. Above the RD50, asphyxiation was first seen intermittently between periods of normal breathing, but was continuous at concentrations that approached lethal levels (i.e., 100 ppm).” The highest concentration of HCN that did not result in death during a 30-min exposure of these mice (100 ppm) was also added to Table 5–5.
Weedon et al. (1940) exposed groups of four mice to HCN at 16 ppm for 16 h. No deaths occurred and mice appeared normal during the exposure. One mouse was autopsied; the organs were described as normal. Mice survived a 24-h exposure to 30 ppm (Pryor et al. 1975).
Sakurai (1989) measured incapacitation times for groups of eight female Jcl ICR mice inhaling various HCN concentrations. HCN was introduced into the exposure chamber from a pressurized tank; chamber concentrations were determined by a “gas detecting tube method.” Animals were placed in rotating cages during the exposures, and incapacitation time was recorded by an electrical signal emitted from the rotating cage at every half rotation. Apnea times were assessed by visual observation. Lack of movement for 5 min was defined as the incapacitation time. The data were graphed, and incapacitation times and concentrations of 5 min, 123.5 ppm; 10 min, 74.4 ppm; 20 min, 50.0 ppm; and 30 min, 41.7 ppm were determined.
3.2.5. Rabbits
Exposure of 24 male Danish rabbits at 0.5 ppm HCN for 4 wk produced no microscopically detectable changes in the lung parenchyma, pulmonary arteries, coronary arteries, or aorta (Hugod 1979).
3.3. Developmental and Reproductive Effects
No information regarding developmental and reproductive effects of HCN in animals via the inhalation route was located in the available literature. The teratogenic potential of inorganic cyanide was studied by infusing sodium cyanide to pregnant golden hamsters between gestation days 6 and 9 (Doherty et al. 1982). Anomalies were induced only at tested doses of 0.126 to 0.1295 mmol/kg/h because preliminary tests had shown that a dose of 0.125 mmol/kg/h did not produce anomalies, and a dose of 0.133 mmol/kg/h produced 100% resorptions. Maternal signs of toxicity were observed after 36 to 48 h, at which time the doses administered by infusion were 30 to 40 times the subcutaneous LD50. This range of doses produced high incidences of congenital malformations and resorptions. The most common anomalies were neural tube defects including encephalocoele and exencephaly. Fetal crown-rump length was significantly reduced in the offspring of treated dams. Maternal toxicity did not correlate with the incidence of anomalies in the offspring. Simultaneous subcutaneous infusion of thiosulfate protected against the teratogenic effects of cyanide. Signs of cyanide poisoning appear if detoxification occurs at a slower rate than absorption (90% of an acute lethal dose of cyanide can be detoxified in an hour when given to guinea pigs by slow infusion). Because signs of maternal toxicity did not appear for 36 to 48 h, the authors suggested that the rate at which sulfur in the form of thiosulfate, cystine, or cysteine became available for cyanide detoxification was the critical step. In addition to sodium cyanide, aliphatic nitriles and cyanogenic glycosides have been demonstrated to be teratogenic to golden hamsters by the oral and inhalation routes (Willhite 1981, 1982; Willhite and Smith 1981; Willhite et al. 1981; Frakes et al. 1985, 1986a,b). The teratogenic activities were attributed to the cyanide released through metabolism of the parent compounds; in each case, developmental toxicity was observed only at doses also inducing signs of maternal cyanide intoxication.
3.4. Genotoxicity
No information regarding the genotoxicity of HCN in animals was located in the available literature. Studies that addressed genotoxicity from other forms of cyanide were reviewed in ATSDR (1997). In those studies, cyanide in the form of potassium cyanide tested negative in Salmonella typhimurium strains TA1535, TA1537, TA1538, TA98, TA10, TA97, and TA102; one study gave positive results with strain TA100. Sodium cyanide gave negative results in several strains of S. typhimurium. Potassium cyanide also tested negative in the DNA repair test in Escherichia coli and in an in vivo testicular DNA synthesis inhibition test with the mouse.
3.5. Chronic Toxicity and Carcinogenicity
No information regarding the carcinogenicity of HCN in animals via the inhalation route was located in the available literature. In a 2-y feeding study, ten male and ten female rats were administered food fumigated with HCN at each of two concentrations (Howard and Hanzal 1955). The average daily concentrations were 73 and 183 mg CN/kg diet. Based on food consumption, body weight, and concentrations at the beginning and end of each feed preparation period, estimated doses were 4.3 and 10.8 mg CN/kg body weight per day. There were no treatment-related effects on body weight and no clinical signs or histopathologic lesions attributable to cyanide ingestion. In a review of feeding studies by the U.S. Environmental Protection Agency (EPA) (1993), 10.8 mg/kg/d (11.2 mg/kg/d as HCN), in the study by Howard and Hanzal (1955), was identified as the highest NOAEL.
3.6. Summary
Lethality data were available for the rat, mouse, and rabbit for exposure periods of 10 s (rat) to 12 h (mouse). Five-minute LC50 values ranged from 323 ppm (mouse) to 503 ppm (rat). Thirty-minute LC50 values ranged from 166 ppm for the mouse to an average of 177 ppm for the rat. The average 1-h LC50 value for the rat was 134 ppm. The LC50 values tend to be similar for the mouse and rat, and the mouse was slightly more sensitive in accordance with its slightly smaller body size and higher relative respiratory rate. Sublethal effects were characterized by incapacitation (or loss of consciousness) and changes in respiratory or cardiac parameters. Exposures causing little to no effect were: monkey, 60 ppm for 30 min—slight changes in EEGs; rat, 200 ppm for 12.5 min—changes in cardiac-released blood enzymes; rat, 55 ppm for 30 min—changes in pulmonary parameters; and mouse, 63 ppm for 30 min—50% decrease in respiratory rate. No information on developmental and reproductive effects, genotoxicity, or carcinogenicity by the inhalation route was located in the available literature. Genotoxicity studies with cyanide salts were generally negative, and no cancers were induced in rats in a 2-y feeding study with HCN.
4. SPECIAL CONSIDERATIONS
4.1. Metabolism and Disposition
HCN is miscible with water and is taken up by the moist respiratory passages. Retention levels of HCN in the nose and lung of human subjects were measured by Landahl and Herrmann (1950) while the subjects inhaled 0.5 to 20 ppm. HCN was delivered to the nose via a mask; the sample was drawn through the nose and out of the mouth while the subject held his breath. Using this procedure, the percentage retained in the nasal passages ranged from 13% to 23%. The percentage retained by the lung when inhaling through the mouth (no mask) ranged from 39% to 77%. The average exposure time was 1 min.
HCN in the blood is almost completely contained in the red blood cells where it is bound to methemoglobin. Immediately after infusion of sodium nitroprusside into patients, 98.4% of the blood cyanide was found in the red blood cells (Vesey et al. 1976). At normal physiological levels of body methemoglobin (0.25% to 1% of the hemoglobin), a human adult can bind about 10 mg of HCN (Schulz 1984).
HCN is detoxified to thiocyanate (SCN−) by the mitochondrial enzyme rhodanese; rhodanese catalyzes the transfer of sulfur from thiosulfate to cyanide to yield thiocyanate, which is relatively nontoxic (Smith 1996). The rate of detoxification of HCN in humans is about 1 μg/kg/min (Schulz 1984) or 4.2 mg/h, which, the author states, is considerably slower than in small rodents. This information resulted from reports of the therapeutic use of sodium nitroprusside to control hypertension. Rhodanese is present in the liver and skeletal muscle of mammalian species as well as in the nasal epithelium. The mitochondria of the nasal and olfactory mucosa of the rat contain nearly seven times as much rhodanese as the liver (Dahl 1989). The enzyme rhodanese is present to a large excess in the human body relative to its substrates (Schulz 1984). This enzyme demonstrates zero-order kinetics, and the limiting factor in the detoxification of HCN is thiosulphate. However, other sulfur-containing substrates, such as cystine and cysteine, can also serve as sulfur donors. Other enzymes, such as 3-mercapto-pyruvate sulfur transferase, can convert cyanide to thiocyanate (ATSDR 1997; NRC 2000). Thiocyanate is eliminated in the urine.
Venous blood levels of cyanide reached a steady state (mean value, 200 μg/100 mL) within 10 min of exposure of cynomolgus monkeys at 100–156 ppm (Purser et al. 1984). The blood level stayed constant during the remainder of the 30-min exposure, during which time the animals lost consciousness; the blood level remained the same for 1 h after exposure, even though the monkeys recovered consciousness within 10 min. The mean concentration of whole blood cyanide in rabbits that died following inhalation exposure was 170 μg/100 mL; the mean plasma concentration was 48 μg/100 mL (Ballantyne 1983).
Plasma levels of cyanide in unexposed, healthy adults average 0 to 10.7 μg/100 mL (mean, 4.8 μg/100 mL) (Feldstein and Klendshoj 1954). Following mild exposures to cyanide, plasma levels return to this normal range within 4 to 8 h after cessation of exposure; the half-life for the conversion of cyanide to thiocyanate from a nonlethal dose in humans was between 20 min and 1 h.
Although Feldstein and Klendshoj (1954) reported plasma levels of cyanide, most data available are for whole blood. Average whole blood values for cyanide are as follows: nonsmokers, 1.6μg/100 mL; smokers, 4.1 μg/100 mL; and nitroprusside therapy, 5 to 50 μg/100 mL (Tietz 1986). These data can be compared with the whole blood values measured in several studies, including the study of Aitken et al. (1977) in which patients were infused with nitroprusside solutions to induce hypotension during intracranial surgery (see Box 1–1). In the Chandra et al. (1980) study, blood cyanide levels of up to 220 μg/100 mL appear excessively high in light of the low measured exposures. Snodgrass (1996) states that blood cyanide greater than 20 μg/100 mL may be associated with acute signs of cyanide poisoning, and deaths occur after blood cyanide reaches 100 μg/100 mL. As noted by Aitken (1977), metabolic acidosis occurred in patients at blood cyanide levels of ≥90 μg/100 mL.
BOX 1–1
Whole Blood Levels of Cyanide in Monitoring and Nitroprusside Therapy Studies. Leeser et al. (1990) Chandra et al. (1980)
It should be noted that HCN can be absorbed through the skin. For this reason, ACGIH (1996) and NIOSH (1997) guidelines carry a skin notation. Drinker (1931) cites the case of three men protected with gas masks in an atmosphere of 2% (20,000 ppm) HCN. After 8 or 10 min the men felt symptoms of marked dizziness, weakness, and throbbing pulse. They left the chamber just before collapse. For several hours after the exposure they experienced weakness, high pulse rate, and headache. They were incapacitated for several days, followed by complete recovery. Based on exposure to several cyanide salts, the dermal LD50 in rabbits was calculated to be 6.7 mg CN−/kg (Ballantyne 1983).
4.2. Mechanism of Toxicity
HCN is a systemic poison that acts on the central nervous system. HCN interrupts cellular respiration by blocking electron transfer from cytochrome oxidase to oxygen. Tissue oxygen levels rise, resulting in increased tissue oxygen tension and decreased unloading for oxyhemoglobin. As a consequence, oxidative metabolism may slow to a point where it cannot meet metabolic demands. This is particularly critical in the brainstem nuclei where lack of an energy source results in central respiratory arrest and death. Cyanide can inhibit many other enzymes, particularly those that contain iron or copper, but cytochrome oxidase appears to be the most sensitive enzyme. Cyanide also stimulates the chemoreceptors of the carotid and aortic bodies to produce a brief period of hyperpnea. Cardiac irregularities may occur, but death is due to respiratory arrest (Hartung 1994; Smith 1996). Brain lesions have been associated with exposure of animals to high concentrations of HCN (ATSDR 1997).
Wexler et al. (1947) studied the effect of intravenously administered sodium cyanide on the electrocardiogram of 16 soldiers. A dose of 0.15 to 0.2 mg/kg (HCN at 0.06–0.11 mg/kg) was chosen based on the known inability of 0.11 mg/kg to stimulate respiration during medical tests (a dose of 0.11 mg of sodium cyanide per kilogram of body weight is used to determine arm-tocarotid blood circulation time). The electrocardiograms of 15 of the 16 men revealed a sinus pause (without auricular activity), which persisted for 0.88 to 4.2 s. The sinus pause immediately preceded or accompanied respiratory stimulation. The pause was followed by marked sinus irregularity, a slowing of the heart rate for a few seconds to 2 min, followed by a gradual acceleration to rates above the baseline level. Baseline heart rate and rhythm were generally restored within 3 min. There was a lesser effect on the sixteenth subject. According to AIHA (2000), this dose is equivalent to inhaling 10 ppm for 1 h.
4.3. Structure-Activity Relationships
No structure-activity relationships were applicable for establishing AEGLs for HCN. It has been observed that the signs of intoxication associated with excessive exposure to HCN and with certain aliphatic nitriles are similar. While the toxic concentrations of acrylonitrile are similar to HCN when compared on the basis of cyanide content (Dudley et al. 1942), the time course of aliphatic nitrile intoxication is different. The authors also observed that dogs are more susceptible to acrylonitrile than monkeys, but repeated exposures to acrylonitrile were more toxic to monkeys than to rats, guinea pigs, or rabbits.
4.4. Other Relevant Information
4.4.1. Species Differences
Lethal concentrations are relatively similar for various animal species and humans (Hartung 1994), with the monkey and goat being the least sensitive, according to Barcroft (1931). Barcroft (1931) reports relative species sensitivity as determined by time to death (in minutes) at a concentration of 1,000 mg/L (910 ppm): dog, 0.8; mouse, cat, and rabbit, 1.0; rat and guinea pig, 2.0; goat, 3.0; and monkey, 3.5. He reported that monkeys (two monkeys per exposure) were only beginning to show signs of unsteadiness when the dogs (two dogs per exposure) died. Also, Barcroft's study (1931) with one human subject and one dog tends to indicate that dogs are much more sensitive to the effects of HCN than humans. Barcroft notes that body size and respiration rate influence the rapidity of effect, small, rapidly respiring animals succumbing first, but he also notes that there are exceptions to the body size effect (i.e., the goat was much less sensitive than the dog). Bancroft's pre-1970 animal studies were not cited in Section 3.1 because time to death is not useful in determining exposure concentration-duration relationships but is useful for determining relative species sensitivity.
Relative to body weight, humans have a much lower respiratory rate and cardiac output than rodents. These are the two primary determinants of systemic uptake of volatile chemicals. Therefore, at similar nominal concentrations, rodents absorb substantially more cyanide than primates. From a pharmacokinetic view, lower hepatic rhodanese levels in primates will not be significant at high, acute HCN exposures. It should be noted that Barcroft's subject withstood a 1 min and 31 s exposure at approximately 500 to 625 ppm without immediate effects (Barcroft 1931), whereas mice suffer asphyxia during a 2 min exposure at 500 ppm (Matijak-Schaper and Alarie 1982). Compared with rodents, the respiratory tracts of humans and monkeys are more similar in gross anatomy, the amount and distribution of types of respiratory epithelium, and airflow patterns (Barrow 1986; Jones et al. 1996).
In the rat and mouse studies by Higgins et al. (1972) and the rat and rabbit studies by Ballantyne (1983), LC50 values differed by less than a factor of two (1.5). All of the 30-min LC50 values summarized in Table 5–4 range from 157 to 200 ppm (rat, mouse, and rabbit and excluding the restrained rats in the study by Levin et al. [1987]). The 1-h LC50 values range from 120 ppm to 144 ppm (data for rat only). The LC30 for the rat at 6 h was 68 ppm. The LC30 and LC50 values are presented graphically in Figure 5–1. The concentrations for the rat are means for the respective time intervals. As can be seen in Figure 5–1, the concentration-time curve is steep, particularly at the shorter time intervals.
FIGURE 5–1
Lethality values for three species of animals. All values are LC50 values except the data point for the rat at 360 min, which is an LC30.
Species differences are recognized in the activity of rhodanese; sheep have relatively high levels of activity and dogs have relatively low levels (Aminlari and Gilanpour 1991). Himwich and Saunders (1948) assayed tissues from several animal species for their ability to produce thiocyanate from cyanide. Activity was generally highest in liver tissue. Rats had the highest levels, dogs had the lowest levels, and rhesus monkeys and rabbits had intermediate levels. Liver and kidney rhodanese activity was two to three times higher in rats and hamsters than in rabbits and female beagles (Drawbaugh and Marrs 1987). The authors point out that in acute exposures at high concentrations, the normal low levels of rhodanese present in tissues would not allow time for substantial detoxification, and other pharmacokinetic considerations may be important in the outcome of acute poisonings.
4.4.2. Susceptible Populations
According to ATSDR (1997), reasons that populations may be more susceptible to the effects of HCN include genetic makeup, age, health and nutritional status, and exposure to other substances. A number of dietary deficiencies, such as vitamin B12 deficiency, may predispose individuals to higher risk for cyanide-associated neuropathies. For example, in tropical areas where cassava is the primary dietary staple, women and children appeared to be more susceptible than adult males to the neurological effects of metabolically liberated cyanide (generated by gut flora from cyanogenic glycosides). These differential responses are observed after repeated ingestion of cyanogenic glycoside-containing foods (e.g., cassava), usually due to the shortage of other dietary staples, particularly those high in protein. No specific information was located on differences in toxicity, metabolism, and/or detoxification between adults and children or between healthy and nutritionally deficient humans following inhalation of HCN.
As noted in Section 4.4.1, the enzyme rhodanese is present to a large excess in the human body relative to its substrates, thus demonstrating zeroorder kinetics (Schulz 1984). This enzyme is functional in newborns, although, in newborns, thiosulphate may be a limiting factor in cyanide detoxification (Schulz and Roth 1982).
Fitzgerald (1954) injected newborn mice (less than 12 h old) and adult mice subcutaneously with sodium cyanide (NaCN). The threshold for lethality was the same in newborn and adult male and female male mice, NaCN at 2 mg/kg. The dose-response curve for neonatal mice was much steeper than for adult mice, which resulted in a lower LC50 value. The LC50 for adult male mice was approximately 5 mg/kg; for female mice it was 3.5 to 3.7 mg/kg; and for neonatal mice it was between 2.0 and 2.5 mg/kg. On the basis of the threshold for lethality, newborn and adult mice were equally sensitive to HCN, but on the basis of LC50 values, newborn mice were approximately two to three times more sensitive than adult male mice.
Individuals with high blood pressure might be considered a susceptible population. Schulz et al. (1982) reported on the infusion of 70 patients, ages 17 to 78, with nitroprusside solutions to lower blood pressure. Administration of nitroprusside with or without thiosulfate continued for several hours to several days, apparently without adverse symptoms. Schulz (1984) states that at 150 to 250 μmol/L of “erythrocyte concentrate” headaches, palpitations, and hyperventilation occur. Unfortunately, blood cyanide levels were expressed in terms of erythrocyte concentrate and could not be compared directly with the data in Section 4.1.
4.4.3. Concentration-Exposure Duration Relationship
When data are lacking for desired exposure times, scaling across time may be based on the relationship between concentration and exposure duration (Cn ×t=k) when a common end point is used (ten Berge et al. 1986). The end points for HCN are incapacitation and lethality. Regression analysis of the data of Sakurai (1989), using incapacitation concentrations for mice for the exposure durations of 5, 10, 20, and 30 min, results in a value for n of 1.6. Regression analysis of the incapacitation data of Purser et al. (1984) for monkeys for the time period of 8 to 19 min results in a value for n of 2.1 (Appendix A, Figure A-1). These studies were of relatively short duration.
FIGURE A-1
Regression line for incapacitation in monkeys (data of Purser et al. [1984])
Several lethality studies conducted over various exposure durations were available for calculation of concentration-exposure duration relationships. Using the animal lethality data of Barcroft (1931), ten Berge et al. (1986) calculated a mean value of 2.7 for n for six species of animals (range, 1.6 to 4.3). The value for the monkey was 1.9 and the value for the rat was 1.6. Using rat and mouse LC50 data sets and exposure times of 5 to 60 min, Hilado and Cumming (1978) calculated an n value of 2. These data indicate a mean n value of 2. Additional data sets were available for the calculation of n values in the present document. Regression analysis of the rat lethality data by E.I. du Pont de Nemours (1981) for exposure durations of 5, 15, 30, and 60 min results in an n value of 2.6 (Appendix A, Figure A-2), and regression analysis of the rat lethality data of Ballantyne (1983), for the exposure durations of 5, 30, and 60 min, results in an n value of 2.1 (data not graphed).
FIGURE A-2
Regression Line for LC50 values in rats (dta of E.I du Pont de Nemours [1981])
It should be noted that extrapolation of the rat 1-h LC50 value of 139 ppm of E.I. du Pont de Nemours (1981) to 6 h (using C2.6×t=k) results in a value of 70 ppm, which is similar to the rat LC30value of Blank (1983), 68 ppm, illustrated in Figure 5–1. Similar results from two different studies support the n value of 2.6 for extrapolation across time in lethality studies with the rat.
4.4.4. Concurrent Exposure Issues
Because many materials release HCN when burned, the combined toxicity of HCN and smoke components—carbon monoxide, carbon dioxide, nitrogen dioxide—have been studied. Combination experiments with fire gases showed that the effects of carbon monoxide and HCN are additive, and a combination of 5% carbon dioxide in HCN decreased the LC50 of HCN for rats (Levin et al. 1987). In 5-min exposures with rats and mice, Higgins et al. (1972) found no measurable interaction between carbon monoxide and HCN. These studies suggest a range of effects, including additive effects, for combinations of gases that may be formed during combustion.
5. DATA ANALYSIS FOR AEGL-1
5.1. Human Data Relevant to AEGL-1
The odor threshold, 0.58 ppm to 5.0 ppm (Amoore and Hautala 1983; Ruth 1986) is low compared with irritant or toxic concentrations. No acute exposures were located resulting in mild effects in humans. Three monitoring studies, involving no symptoms to mild symptoms during chronic occupational exposures of adult males, are relevant to development of AEGL-1 values. The symptoms and blood concentrations of cyanide in the monitoring study of Chandra et al. (1980) indicate that the workers may have been exposed at higher atmospheric concentrations than those reported.
Mean concentrations of cyanide in the breathing zone of workers (all nonsmokers) in an electroplating area of three factories were 6, 8, and 10 ppm (range, 4.2–12.4 ppm) (El Ghawabi et al. 1975). Employment ranged from 5 to 15 y. Complaints of headache, weakness, and changes in taste and smell were reported by approximately 80% of the workers; incidences were much higher than in a matched control group. Irritation of the throat, vomiting, and effort dyspnea were commonly reported, and lachrimation and precordial pain were reported relatively less frequently. Two workers in the factory with the highest exposures suffered from psychotic episodes during the survey. Twenty of the 36 workers had thyroid enlargement to a mild or moderate degree. Air cyanide concentrations and exposure durations were not linked to specific symptoms. Mean levels of thiocyanates in the urine correlated with air concentrations of cyanide. Although the sample size was small, 36 workers, the study used 20 well-matched controls and a biological index of exposure (urinary thiocyanate). An NRC subcommittee concluded from this study that 1-h exposures at 8 ppm might produce mild headache in healthy adults (NRC 2000).
The Leeser et al. (1990) study was a controlled study with comprehensive medical examinations. In this study, presumably healthy workers were exposed to geometric mean HCN concentrations up to 1 ppm (range, 0.01–3.3 ppm) determined by personal monitoring in the work areas. Concentrations in the atmosphere of the plant ranged up to 6 ppm during the fall of the year, as indicated by Draeger pump tests or static monitors. It is not clear that the geometric mean concentrations include the later, higher values, as ranges during the spring were reported to be up to only 3.3 ppm. Higher blood cyanide levels were correlated with the higher exposure levels in the fall of the year. The results of clinical histories and medical examinations showed no differences to only minor differences for a variety of parameters between the HCN workers and a matched control group.
Medical questionnaires were not given in the Grabois (1954) study. However, both NIOSH (1976) and ACGIH (1996) reviewed the study. NIOSH (1976) identified 5 ppm as a no-effect concentration using the data for the five plants presented by Grabois (1954). Similar exposures were reported in the studies of Hardy et al. (1950) and Maehly and Swensson (1970).
5.2. Animal Data Relevant to AEGL-1
Animal studies that addressed sensory irritation or mild effects were not clearly distinguishable from those that addressed more severe effects.
5.3. Derivation of AEGL-1
The AEGL-1 is based on monitoring studies in which the preponderance of data as a weight-of-evidence consideration indicates that an 8-h exposure to 1 ppm would be without adverse effects for the general population. El Ghawabi et al. (1975) reported symptoms such as headache, weakness, changes in taste and smell, irritation of the throat, vomiting, and effort dyspnea in three electroplating plants in which mean concentrations of HCN were 6, 8, and 10 ppm, but the authors failed to relate symptoms to air concentrations. It should be noted that 20 of the 36 workers in the El Ghawabi et al. (1975) study had thyroid enlargement, which is characteristically observed in cases of chronic cyanide exposure and may have been responsible for some of the symptoms. An NRC subcommittee, in evaluating the El Ghawabi et al. (1975) data, concluded that the average concentration of 8 ppm in the three plants would likely produce no more than mild headache, which would be acceptable for a 1-h exposure of healthy adults. In the monitoring study of Leeser et al. (1990), chronic exposure of 63 workers in a cyanide salt production plant to geometric mean concentrations up to approximately 1 ppm and possible excursions up to 6 ppm during part of the year produced no clear exposure-related symptoms. According to NIOSH (1976), chronic exposure of workers to 5 ppm while processing apricot kernels in the monitoring study of Grabois (1954) was without effect. Additional monitoring studies with mean exposures to 5 ppm failed to report adverse health effects (Hardy et al. 1950; Maehly and Swensson 1970). It is unlikely that the population of workers in these and additional monitoring studies represent only healthy individuals.
The AEGL-1 was derived from a consideration of the dose-response data, which were obtained from all of the monitoring studies and subsequently time scaled to the shorter exposure durations. Although the exposures were of chronic duration in the monitoring studies, they represent the best available human data. Symptoms observed during chronic exposures should represent the greatest potential response. An 8-h exposure duration was selected as the basis for AEGL development.
Mild headache is a symptom of exposure that meets the definition of an AEGL-1. Dividing the 8-h concentration of 5 ppm of the Grabois (1954), Hardy et al. (1950), or Maehly and Swensson (1970) study by an intraspecies uncertainty factor (UF) of 3 or dividing the 1-h concentration of 8 ppm of the El Ghawabi et al. (1975) study by an intraspecies UF of 3 results in very similar AEGL-1 values. The resulting 8-h value of 1.7 ppm is also similar to the 8-h no-effect concentration of 1 ppm in the Leeser et al. (1990) study, where no UF was applied. UFs are generally applied to the highest NOAELs or lowest LOAELs. A UF was not applied to the Leeser et al. (1990) study because it was the lowest NOAEL. No specific susceptible populations were identified during numerous occupational monitoring studies or during the clinical use of nitroprusside solutions to control hypertension. Thus, potential differences in susceptibility among humans are not expected to exceed 3-fold. All individuals, including infants, possess large amounts of the cyanide detoxifying enzyme rhodanese (as well as other detoxifying enzymes) and normally have adequate amounts of sulfur-containing compounds.
The 8-h no-effect mean geometric concentration of 1 ppm (with excursions up to 6 ppm) from the Leeser et al. (1990) study was used as the basis for time scaling the AEGL-1 values. This study was chosen because it was well conducted: all workers had full medical examinations and routine blood tests, including measurements of blood cyanide and carboxyhemoglobin. Atmospheric HCN concentrations were monitored in the plant several times during the year. Because of the extrapolation from a long-term exposure, the 8-h value of 1 ppm was time scaled to the other exposure durations using the relationship C3×t=k where k=480 ppm3·min. In order to stay below the highest measured concentration from personal samplers, at 3.3 ppm in the Leeser et al. (1990) study, the 10-min value was set equal to the 30-min value. Calculations are in Appendix B, and values appear in Table 5–6 below.
TABLE 5–6
AEGL-1 Values for Hydrogen Cyanide.
6. DATA ANALYSIS FOR AEGL-2
6.1. Human Data Relevant to AEGL-2
As noted above for the AEGL-1, chronic occupational exposure of adult males to >10 ppm produced symptoms of headache, weakness, changes in taste and smell, irritation of the throat, vomiting, and effort dyspnea (El Ghawabi et al. 1975; NIOSH 1976; Blanc et al. 1985). For a few individuals, chronic exposures occasionally produced more serious adverse effects, such as fainting and psychotic episodes. There was no evidence that these symptoms occurred after one exposure. A concentration of ≥25 ppm for 1 h resulted in numbness, weakness, vertigo, nausea, rapid pulse, and flushing of the face (Parmenter 1926). Only one individual was involved, and neither the exposure duration nor the concentration were measured.
6.2. Animal Data Relevant to AEGL-2
Several animal studies listed in Table 5–5 describe effects at concentrations below those causing incapacitation or unconsciousness. These 30-min studies are as follows: monkey, 60 ppm (slight CNS effects) (Purser 1984); rat, 55 ppm (changes in lung dynamics and phospholipids) (Bhattacharya et al. 1984); and mouse, 63 ppm (respiratory depression of 50%) (Matijak-Schaper and Alarie 1982). From the description given by Matijak-Schaper and Alarie (1982), the concentration of 63 ppm for 30 min appears to be the threshold for a breathing pattern characteristic of asphyxiation. The effects in these three studies are reversible and do not impair the ability to escape, but they can be considered close to the threshold for such effects. Incapacitation in monkeys occurs at higher concentrations (80–150 ppm) (Purser et al. 1984). The 24-h exposure of mice at 30 ppm (Pryor et al. 1975), resulting in lung congestion, is also relevant to the definition of AEGL-2. The data of Sakurai (1989), incapacitation in mice inhaling 41.7 ppm in rotating cages for 30 min, appear low compared with the other studies and were not considered.
6.3. Derivation of AEGL-2
Because the human exposure concentrations are less reliable than the experimental animal data, the animal data were used in the derivation of the AEGL-2 values. The study chosen for the AEGL-2 derivation was the study by Purser (1984) with the monkey, because it was well conducted and used an appropriate species (compared with the rodent, the respiratory tracts of humans and monkey are more similar in anatomy, the amount and distribution of types of respiratory epithelia, and airflow pattern). This concentration was 60 ppm for 30 min. Although this end point (a slight depressive effect on the central nervous system as evidenced by a change in brain-wave activity near the end of the exposure) was a NOAEL for the definition of an AEGL-2, it was chosen because the next higher experimental concentration resulted in severe adverse effects of incapacitation, unconsciousness, and possibly death. The Barcroft (1931) lethality and incapacitation study has shown that the monkey is less sensitive to the respiratory and central nervous system effects of HCN than the rat and mouse (by factors of 1.75 and 3, respectively), and the adult human is less sensitive than the dog. The differences in sensitivity were based, at least partially, on the more rapid respiratory rates and greater cyanide uptake of rodents and the dog compared with humans and the monkey.
Because the respiratory tracts of humans and monkeys are more similar than those of humans and rodents, because uptake is more rapid in the monkey than in humans, and because both species have been shown to be relatively insensitive to the incapacitative and lethal effects of HCN (but at the same time, species susceptibilities to lethal effects do not differ by more than a factor of 1.5), an interspecies UF of 2 was applied. Human (adult) accidental and occupational exposures (El Ghawabi et al. 1975) indicate that there are individual differences in sensitivity to HCN, as evidenced by symptoms following chronic exposures, but the magnitude of these differences does not appear to be great. These studies and the clinical use of nitroprusside solutions to control hypertension do not demonstrate a susceptible population. The detoxifying enzyme rhodanese is functional in all individuals, including newborns. Therefore, a UF of 3 was applied to account for potential differences in human susceptibility. For the concentration-exposure duration relationship, the mean value for n of 2.0 for the monkey was calculated from two data sets involving incapacitation (2.1) and lethality (1.9) (Section 4.4.3). The 30-min exposure value of 60 ppm was divided by a total UF of 6 and scaled across time using the Cn×t=k, where n=2 and k=3,000 ppm2·min. Values appear in Table 5–7 below, and calculations are in Appendix B.
TABLE 5–7
AEGL-2 Values for Hydrogen Cyanide.
The safety of the values is supported by the data of Grabois (1954), in which occupational exposures ranged up to 17 ppm, and two additional animal studies. The 30-min exposure of rats at 55 ppm (Bhattacharya et al. 1984), when divided by a total UF of 6 (2 for interspecies and 3 for intraspecies), results in a 30-min AEGL-2 of 9.2 ppm. The described effects of changes in lung dynamics and lung phospholipids are not irreversible or long-lasting. Mice experienced a decrease of 50% in respiratory rate when inhaling 63 ppm for 30 min but did not lose consciousness (Matijak-Schaper and Alarie 1982; Alarie 1997). Dividing by a total UF of 6 results in a 30-min AEGL-2 value of 11 ppm.
7. DATA ANALYSIS FOR AEGL-3
7.1. Human Data Relevant to AEGL-3
No human studies of sufficient exposure duration with measured concentrations producing irreversible or life-threatening effects were located in the available literature. However, the data of Barcroft (1931), a 1.5-min exposure at 500–625 ppm, and Bonsall (1984), a 6-min exposure at approximately 450 ppm, with recovery from symptoms and effects, can be considered short-term upper limits for healthy adults.
7.2. Animal Data Relevant to AEGL-3
LC01 values for four time periods were provided by E.I. du Pont de Nemours (1981) for the rat. They are as follows: 5 min, 283 ppm; 15 min, 138 ppm; 30 min, 127 ppm; and 60 min, 88 ppm. Ballantyne (1983) used several concentrations and exposure durations but did not provide the actual concentrations; therefore, an LC01 could not be calculated. Matijak-Schaper and Alarie (1982) reported no deaths in mice inhaling HCN at 100 ppm for 30 min. Mice inhaling HCN at 30 ppm for 24 h showed signs of lung congestion (Pryor et al. 1975).
7.3. Derivation of AEGL-3
The 15- and 30-min and 1-h LC01 values of 138, 127, and 88 ppm, respectively, provided by E.I. du Pont de Nemours (1981) for the rat were used to derive the AEGL-3 values. Lethal concentrations are very similar for various animal species (Table 5–4), and Barcroft (1931) has shown that man and the monkey are less sensitive to the effects of HCN than are the rat and dog, a conclusion based at least partially on relative respiratory rates. Relative to body weight, humans have a much lower respiratory rate and cardiac output than rodents. These are the primary determinants of systemic uptake of volatile chemicals. Thus, at similar exposure concentrations, rodents will absorb substantially more cyanide than primates. Lower rhodanese activity levels in primates will not be significant at high, acute HCN exposure levels. These factors might argue for use of an interspecies UF of 1. However, an interspecies UF was applied because of the high acute toxicity and rapid action of HCN. Because LC50 values among animal species differed by less than a factor of 2, an interspecies UF of 2 was applied. Human accidental and occupational exposures indicate that there are individual differences in sensitivity to HCN, but the magnitude of these differences does not appear to be great. No specific data on susceptible populations were located in numerous published monitoring studies or during the clinical use of nitroprusside solutions to control hypertension. The detoxifying enzyme rhodanese, as well as other enzymes, is functional in all individuals, including newborns. Therefore, a UF of 3 was applied to protect susceptible individuals. The concentration-exposure duration relationship for this data set is C2.6×t=k (Section 4.4.3); therefore, the value of 2.6 for n was applied. The 15- and 30-min and 1-h values were divided by a total UF of 6 and the 15-min and 1-h values were scaled across time using the C2.6×t=k relationship (the 15-min value for the 10-min AEGL-3 and the 1-h for the 4- and 8-h AEGL-3 values). Values appear in Table 5–8 and calculations are in Appendix B.
TABLE 5–8
AEGL-3 Values for Hydrogen Cyanide.
The AEGL values are supported by the study of Pryor et al. (1975) with the mouse in which a 24-h exposure at 30 ppm induced pulmonary congestion but was not lethal. The 30 ppm concentration divided by a total UF of 6 and scaled across time from 24 h to 30 min using C2.6×t=k results in a 30-min AEGL-2 of 22 ppm. The AEGL values are also supported by the study of Parmenter (1926) in which an individual potentially exposed at 25–75 ppm for part of a day had severe symptoms but recovered fully. Furthermore, Barcroft's subject withstood a 1.5-min exposure at 500–625 ppm (Barcroft 1931). Time scaling the AEGL-3 values to 1.5 min results in a concentration at 60 ppm, which is less than the actual exposure by a factor of approximately 10.
8. SUMMARY OF AEGLs
8.1. AEGL Values and Toxicity End Points
The AEGL values and toxicity end points are summarized in Table 5–9.
TABLE 5–9
Summary of AEGL Values (ppm [mg/m3]).
8.2. Comparisons with Other Standards and Guidelines
Standards and guidance levels for workplace and community exposures are listed in Table 5–10. The Leeser et al. (1990) study was not available at the time many of these standards and guidelines were developed. The American Industrial Hygiene Association (AIHA 2000) did not derive an ERPG-1 value. The 1-h AEGL-2 and AEGL-3 values are slightly lower than the corresponding 1-h ERPG values. The ERPG-2 value was based on the Wexler et al. (1947) study in which sodium cyanide given intravenously to human volunteers at 0.11 mg/kg caused no deaths or serious injuries. The AIHA suggested that the intravenous dose approximates a 1-h exposure at 10 ppm. Because a bolus dose of cyanide does not take into account metabolism over the 1-h exposure duration, a well-conducted animal study was chosen with an appropriate species, but inter- and intraspecies UFs were applied. The ERPG-3 was based on several animal studies, including Purser (1984), in which concentrations of 45 to 60 ppm resulted in only reversible effects. These studies, and several additional lethality studies, were also reviewed for the AEGL-3.
TABLE 5–10
Extant Standards and Guidelines for Hydrogen Cyanide.
The 1-h AEGL-2 (7.1 ppm) is close to the 1-h Spacecraft Maximum Allowable Concentration (SMAC) of 8 ppm (NRC 2000), and both groups considered available monitoring studies in their derivations. Although the SMAC definition is similar to the AEGL-1 definition, the SMAC applies to healthy adults, whereas the AEGL-2 applies to the general population; therefore, the AEGL-2 value is conservative in comparison with the SMAC. The NRC subcommittee on SMACs used the monitoring data of El Ghawabi et al. (1975) to develop the values. The subcommittee suggested that the average concentration of “8.0 ppm in the three plants would likely produce no more than mild CNS effects (e.g., mild headache), which would be acceptable for 1-hour exposures in a spacecraft.” The subcommittee concluded that it was “likely that the more serious symptoms, such as vomiting, were the result of brief exposures to high HCN concentrations.” Therefore, 8 ppm was identified as the 1-h allowable concentration of HCN. The 24-h SMAC is 4 ppm and the 7-d SMAC is 1 ppm.
The NIOSH immediately dangerous to life and health (IDLH) value (NIOSH 1994) is greater than the 30-min AEGL-3. NIOSH based their recommended exposure limit (REL) on the statement by Flury and Zernik (1931) that 45–54 ppm could be tolerated by man for 0.5 to 1 h without immediate or late effects. Although the Flury and Zernik (1931) data are based on animal studies, NIOSH did not apply a UF.
Both ACGIH (1996) and NIOSH (1999) based their ceiling and short-term exposure limits, respectively, on one of the studies used for development of the AEGL-1. The value for both agency limits of 4.7 ppm was based on symptoms described during chronic exposures of workers in several studies and specifically on El Ghawabi et al. (1975). In 1993, the ACGIH value was reduced from 10 ppm to minimize the potential for irritation to the respiratory tract as well as potential acute and chronic effects of cyanide. The German and Dutch occupational exposure concentrations, analogous to the 8-h ACGIH time-weighted average (TWA), are 4.7 and 10 ppm, respectively. The German maximum workplace concentration (MAK) peak category or ceiling value is two times the MAK; this concentration is of a 5-min maximum duration and must not be exceeded at any time during the work shift.
8.3. Data Adequacy and Research Needs
The data base from animal studies is robust, but there are little definitive data on human exposure concentrations for short exposure durations and no definitive data on differences in susceptibilities among adults or between adults, infants, and children, other than well-understood ventilatory differences in the latter case. Gender and age-related differences in response to chronic cyanogenic glycoside consumption are difficult to interpret due to confounding, marked protein deficiencies in those populations that consume cassava as a major dietary staple (see Section 4.4.2). However, monitoring studies of presumably healthy adults that established no effect and/or minor discomfort concentrations to inhaled cyanide were available to set projected safe levels for the entire population by applying appropriate uncertainty factors (UFs). The metabolism and mechanism of action of cyanide are well understood and identical in all mammalian species. Data were available on concentrations involving lethal and sublethal effects for the monkey, dog, rat, mouse, and rabbit. Exposure durations included those ranging from a few seconds to 24 h. Where different mammalian species were tested in the same study, the results indicated that sensitivity to cyanide toxicity is similar among species, but slight differences may be related to body size, which in turn is related to respiration rate. Thus, establishing safe levels for humans based on small mammalian species adds confidence to the AEGL derivation. Animal studies with different toxicologic end points were available to establish concentration-exposure duration relationships. The extreme toxicity of HCN precludes certain types of tests, including long-term inhalation studies; therefore, genotoxicity, carcinogenicity and developmental and reproductive studies were performed with cyanide salts.
Several studies provided data on blood and urine concentrations of cyanide and thiocyanate following occupational exposures at low concentrations. These values are generally similar to those of smokers who have not been occupationally exposed to HCN. Whole-blood cyanide concentrations during nitroprusside infusion also have been measured and related to symptoms. There are also data on nonlethal oral doses and metabolism rates in humans. Taken together, the data indicate that the HCN AEGL values may be conservative. However, data on infants, children, and the elderly, populations that may be more susceptible to HCN toxicity than healthy adults based on higher respiration rates and slower metabolism, among other factors, are lacking. Furthermore, occupational monitoring data were collected under normal working conditions; stress or physical exertion may be greater under emergency conditions. Because HCN is extremely toxic and the range of human susceptibility is not definitively known, the AEGL derivations make use of appropriate UFs.
9. REFERENCES
- ACGIH (American Conference of Governmental Industrial Hygienists). 1996. Documentation of the Threshold Limit Values and Biological Exposure Indices: Hydrogen cyanide. Seventh ed., ACGIH, Cincinnati, OH.
- ACGIH (American Conference of Governmental Industrial Hygienists). 2000. Threshold Limit Values (TLVs) for Chemical and Physical Agents and Biological Exposure Indices (BEIs). ACGIH, Cincinnati, OH.
- AIHA (American Industrial Hygiene Association). 2000. The AIHA 2000 Emergency Response Planning Guidelines and Workplace Environmental Exposure Level Guidelines Handbook. AIHA, Fairfax VA.
- Aitken, D., D.West, F.Smith, W.Poznanski, J.Cowan, J.Hurtig, E.Peterson, and B.Benoit. Cyanide toxicity following nitroprusside induced hypotension. Can. Anes. Soc. J. 24:651–660. [PubMed: 589503]
- Alarie, Y. 1997. Personal Communication to Sylvia Talmage, Oak Ridge NationalLaboratory, via e-mail, November 20, 1997.
- Aminlari, M., and H.Gilanpour. 1991. Comparative studies on the distribution of rhodanese in different tissues of domestic animals. Comp. Biochem. Physiol. 99B:673–677. [PubMed: 1769215]
- Amoore, J.E. and E.Hautala. 1983. Odor as an aid to chemical safety: Odor thresholds compared with Threshold Limit Values and volatilities for 214 industrial chemicals in air and water dilution. J. Appl. Toxicol. 3:272–290. [PubMed: 6376602]
- ATSDR (Agency for Toxic Substances and Disease Registry). 1997. Toxicological profile for cyanide. U.S. Department of Health and Human Services, Public Health Service, Atlanta, GA.
- Ballantyne, B. 1983. The influence of exposure route and species on the acute lethal toxicity and tissue concentrations of cyanide. In: Developments in the Science and Practice of Toxicology. Elsevier Science Publishers, New York, pp. 583– 586. [PubMed: 6428857]
- Barcroft, J. 1931. The toxicity of atmospheres containing hydrocyanic acid gas. J. Hyg. 31:1–34. [PMC free article: PMC2170589] [PubMed: 20475080]
- Barrow, C.S. 1986. Toxicology of the Nasal Passages. New York: Hemisphere Publishing Corporation.
- Bhattacharya, R., P.Kumar, and A.S.Sachan. 1994. Cyanide induced changes in dynamic pulmonary mechanics in rats. Indian J. Pharmacol. 38:281–284. [PubMed: 7883293]
- Blanc, P., M.Hogan and M.Mallin. 1985. Cyanide intoxication among silver-reclaiming workers. J. Amer. Med. Assoc. 253:367–371. [PubMed: 3965790]
- Blank, T.L. 1983. Inhalation Pilot Study of Hydrogen Cyanide Exposure in Sprague-Dawley Rats. Report No. MSL-2985, Monsanto Company. U.S. EPA OTS Submission 88–920007543.
- Bonsall, J.L. 1984. Survival without sequelae following exposure to 500 mg/m3 of hydrogen cyanide. Human Toxicol. 3:57–60. [PubMed: 6321328]
- Budavari, S., editor; M.J.O'Neil, editor; , A.Smith, editor; , P.E.Heckelman, editor; and J.F.Kinneary, editor. (Eds.). 1996. The Merck Index, 12th ed. Merck & Co., Inc., Rahway, NJ.
- Carson, B.L., H.V.Ellis, B.L.Herndon, E.M.Horn, and L.H.Baker. 1981. Hydrogen Cyanide Health Effects. EPA-460/3–81–026, U.S. EPA, Office of Mobile Source Air pollution Control, Ann Arbor, MI.
- CEH. 2000. CEH Abstract: Hydrogen cyanide. Chemical Economics Handbook, SRI Consulting, retrieved from http://piglet
.sri.com/CIN/June2000. - Chandra, H., B.N.Gupta, S.K.Bhargava, S.H.Clerk and P.N.Mahendra. 1980. Chronic cyanide exposure—A biochemical and industrial hygiene study. J. Anal. Toxicol. 4:161–165. [PubMed: 6257975]
- Dahl, A.R. 1989. The cyanide-metabolizing enzyme rhodanese in rat nasal respiratory and olfactory mucosa. Toxicol. Lett. 45:199–205. [PubMed: 2919401]
- Doherty, P.A., V.H.Ferm, and R.P.Smith. 1982. Congenital malformations induced by infusion of sodium cyanide in the golden hamster. Toxicol. Appl. Pharmacol. 64:456–464. [PubMed: 7135396]
- Drawbaugh, R.B. and T.C.Marrs. 1987. Interspecies differences in rhodanese (thiosulfate sulfurtransferase, EC 2.8.1.1) activity in liver, kidney and plasma. Comp. Biochem. Physiol.86B:307–310. [PubMed: 3105953]
- Drinker, P. 1932. Hydrocyanic acid gas poisoning by absorption through the skin. J. Ind. Hyg. 14:1–2.
- Dudley, H.C., T.R.Sweeney, and J.W.Miller. 1942. Toxicology of acrylonitrile (vinyl cyanide). II. Studies of effects of daily inhalation. J. Ind. Hyg. Toxicol. 24:255–258.
- E.I. du Pont de Nemours and Company. 1981. Inhalation toxicity of common combustion gases. Haskell Laboratory Report No. 238–81. Haskell Laboratory, Newark, DE.
- El Ghawabi, S.H., M.A.Gaafar, A.A.El-Saharti, S.H.Ahmed, K.K.Malash and R. Fares. 1975. Chronic cyanide exposure: A clinical, radioisotope, and laboratory study. Brit. J. Ind. Med.32:215–219. [PMC free article: PMC1008061] [PubMed: 1156569]
- EPA (U.S. Environmental Protection Agency). 1993. Hydrogen cyanide. IRIS online data base, http://www
.epa.gov/ngispgm3/iris. - Feldstein, M. and N.C.Klendshoj. 1954. The determination of cyanide in biologic fluids by microdiffusion analysis. J. Lab. Clin. Med. 44:166–170. [PubMed: 13174934]
- Fitzgerald, L.R. 1954. Effect of injected sodium cyanide on newborn and adult mice. Am. J. Physiol. 17:60–62. [PubMed: 13207387]
- Flury, F. and F.Zernik. 1931. Schadliche gase dampfe, nebel, rauch- und staubarten. Berlin, Germany: Verlag von Julius Springer.
- Frakes, R.A., R.P.Sharma, and C.C.Willhite. 1985. Developmental toxicity of the cyanogenic glycoside linamarin in the golden hamster. Teratology 31:241–246. [PubMed: 3992492]
- Frakes, R.A., R.P.Sharma, and C.C.Willhite. 1986. Comparative metabolism of linamarin and amygdalin in hamsters. Fd. Chem. Toxic. 24:417–420. [PubMed: 3744195]
- Frakes, R.A., R.P.Sharma, C.C.Willhite, and G.Gomex. 1986. Effect of cyanogenic glycosides and protein content in cassava diets on hamster prenatal development. Fund. Appl. Toxicol.7:191–198. [PubMed: 3758536]
- German Research Association (Deutsche Forschungsgemeinschaft). 1999. List of MAK and BAT Values, 1999. Commission for the Investigation of Health Hazards of Chemical Compounds in the Work Area, Report No. 35. Weinheim, Federal Republic of Germany: Wiley VCH.
- Grabois, B. 1954. Monthly Review 33:33; Publication of the Division of Industrial Hygiene, New York Department of Labor, September 1954.
- Hardy, H.L., W.M.Jeffries, M.M.Wasserman, and W.R.Waddell. 1950. Thiocyanate effect following industrial cyanide exposure—report of two cases. New Engl. J. Med. 242:968–972. [PubMed: 15423689]
- Hartung, R. 1994. Cyanides and nitriles. In Patty's Industrial Hygiene and Toxicology, 4th ed., vol. II , Part D. John Wiley & Sons, pp.3119–3172.
- Higgins, E.A., V.Fiorca, A.A.Thomas and H.V.Davis. 1972. Acute toxicity of briefexposures to HF, HCl, NO2 and HCN with and without CO. Fire Technol. 8:120–130.
- Hilado, G.J. and H.J.Cumming. 1978. Short-term LC50 values: An update on available information. Fire Technol. 14:46–50.
- Howard, J.W., and R.F.Hanzal. 1955. Chronic toxicity to rats of food treated with hydrogen cyanide. Agric. Food Chem. 3:325–329.
- HSDB (Hazardous Substances Data Bank). 2000. MEDLARS Online Information Retrieval System, National Library of Medicine, retrieved 9/1/00.
- Hugod, C. 1979. Effect of exposure to 0.5 ppm hydrogen cyanide singly or combined with 200 ppm carbon monoxide and/or 5 ppm nitric oxide on coronary arteries, aorta, pulmonary artery, and lungs in the rabbit. Int. Arch. Occup. Environ. Health 44:13–23. [PubMed: 230157]
- Jones, T.C., editor; , D.L.Dungworth, editor; , and U.Mohr, editor. , eds. 1996. Monographs on Pathology of Laboratory Animals: Respiratory System. New York: Springer Verlag.
- Kimmerle, G. 1974. Aspects and methodology for the evaluation of toxicological parameters during fire exposure. Combust. Toxicol. 1:4–51.
- Kumar, P., M.Das, and A.Kumar. 1991. Health status of workers engaged in heat treatment (case hardening) plant and electroplating at cyanide bath. Indian J. Environ. Prot. 12:179–183.
- Landahl, H.D. and R.G.Herrmann. 1950. Retention of vapors and gases in the human nose and lung. Arch. Indust. Hyg. Occup. Med. 1:36–45. [PubMed: 15419854]
- Leeser, J.E., J.A.Tomenson, and D.D.Bryson. 1990. A cross-sectional study of the health of cyanide salt production workers. Report No. OHS/R/2, ICI Central Toxicology Laboratory, Alderley Park, Maccles field, Cheshire, U.K.
- Levin, B.C., M.Paabo, J.L.Gurman and S.E.Harris. 1987. Effects of exposure to single or multiple combinations of the predominant toxic gases and low oxygen atmospheres produced in fires. Fund. Appl. Toxicol. 9:236–250. [PubMed: 2820822]
- Maehly, A.C. and A.Swensson. 1970. Cyanide and thiocyanate levels in blood and urine of workers with low-grade exposure to cyanide. Int. Arch. Arbeitsmed. 27:195–209. [PubMed: 5490372]
- Matijak-Schaper, M., and Y.Alarie. 1982. Toxicity of carbon monoxide, hydrogen cyanide and low oxygen. Combust. Technol. 9:21–61.
- McNamara, B.P. 1976. Estimates of the toxicity of hydrocyanid acid vapors in man. Edgewood Arsenal Technical Report EB-TR-76023, Department of the Army, August, 1976.
- Ministry of Social Affairs and Employment (SDU Uitgevers). 2000. Nationale MAC (Maximum Allowable Concentration) List, 2000. The Hague, The Netherlands.
- NIOSH (National Institute for Occupational Safety and Health). 1976. Criteria for a Recommended Standard…. Occupational Exposure to Hydrogen Cyanide and Cyanide Salts (NaCN, KCN, and Ca(CN)2). PB 266 230, DHEW (NIOSH) Pub. No. 77–108, U.S. Department of Health, Education, and Welfare; U.S. Government Printing Office, Washington, DC.
- NIOSH (National Institute for Occupational Safety and Health). 1994. Documentation for Immediately Dangerous to Life or Health Concentrations (IDLHS). National Institute for Occupational Safety and Health, Cincinnati, OH; PB94195047, National Technical Information Service, Springfield, VA.
- NIOSH (National Institute for Occupational Safety and Health). 1997. NIOSH Pocket Guide to Chemical Hazards. Publication 94–116, U.S. Department of Health and Human Services; U.S. Government Printing Office, Washington, DC.
- NRC (National Research Council). 1984. Emergency and continuous exposure limits for selected airborne contaminants, Volume 2. Committee on Toxicology, National Research Council, National Academy Press, Washington, DC.
- NRC (National Research Council). 1993. Guidelines for Developing Community Emergency Exposure Levels for Hazardous Substances. Washington, DC: National Academy Press.
- NRC (National Research Council). 2000. Hydrogen cyanide. In Spacecraft Maximum Allowable Concentrations for Selected Airborne Contaminants, Volume 4. Washington, DC; National Academy Press, pp.330–365.
- NRC (National Research Council). 2001. Standing Operating Procedures for Developing Acute Exposure Guideline Levels for Hazardous Chemicals. Washington, DC: National Academy Press.
- O'Flaherty, E.J. and W.C.Thomas. 1962. The cardiotoxicity of hydrogen cyanide as a component of polymer pyrolysis smokes. Toxicol. Appl. Pharmacol. 63:373– 381. [PubMed: 6285553]
- Parmenter, D.C. 1926. Observations on mild cyanide poisoning: Report of a case. J. Indust. Hyg. 8:280–282.
- Peden, N.R., A.Taha, and P.D.McSorley. 1986. Industrial exposure to hydrogen cyanide: Implications for treatment. Br. Med. J. 293:538. [PMC free article: PMC1341311] [PubMed: 3019459]
- Pesce, L.D. 1994. Cyanides: Hydrogen cyanide. In Kirk-Othmer Encyclopedia of Chemical Technology, Vol. 7,4th ed. John Wiley & Sons, New York, pp. 753– 765.
- Pryor, A.J., D.E.Johnson and N.N.Jackson. 1975. Hazards of smoke and toxic gases produced in urban fires. Combust. Toxicol. 2:64–112.
- Purser, D.A. 1984. A bioassay model for testing the incapacitating effects of exposure to combustion product atmospheres using cynomolgus monkeys. J. Fire Sciences 2:20–36.
- Purser, D.A., P.Grimshaw, and K.R.Berrill. 1984. Intoxication by cyanide in fires: A study in monkeys using polyacrylonitrile. Arch. Environ. Health 39:394–400. [PubMed: 6098227]
- Radojicic, B. 1973. Determining thiocyanate in urine of workers exposed to cyanides. Arh. Hig. Rada 24:227–232. [PubMed: 4786702]
- Ruth, J.H. 1986. Odor thresholds and irritation levels of several chemical substances: A review. Amer. Ind. Hyg. Assoc. J. 47:A142–A151. [PubMed: 3706135]
- Sakurai, T. 1989. Toxic gas tests with several pure and mixed gases using mice. J. Fire Sci. 7:22–77.
- Schulz, V. 1984. Clinical pharmacokinetics of nitroprusside, cyanide, thiosulphate and thiocyanate. Clin. Pharmacokin. 9:239–251. [PubMed: 6375932]
- Schulz, V., and B.Roth. 1982. Detoxification of cyanide in a new-born child. Klin. Wochensch. 60:527–528. [PubMed: 7098381]
- Schulz, V., R.Gross, T.Pasch, J.Busse, and G.Loeschcke. 1982. Cyanide toxicity of sodium nitroprusside in therapeutic use with and without sodium thiosulphate. Klin. Wochensch.60:1393–1400. [PubMed: 7176461]
- Singh, B.M., N.Coles, and P.Lewis. 1989. The metabolic effects of fatal cyanide poisoning. Postgrad. Med. J. 65:923–925. [PMC free article: PMC2429567] [PubMed: 2616434]
- Smith, R.P. 1996. Toxic responses of the blood. In: Casarett & Doull's Toxicology: The Basic Science of Poisons, 5th Ed., McGraw-Hill, New York, pp.350–351.
- Snodgrass, W.R.. 1996. Clinical toxicology. In: Casarett & Doull's Toxicology: The Basic Science of Poisons, 5th Ed., McGraw-Hill, New York.
- ten Berge, W.F., A.Zwart and L.M.Appleman. 1986. Concentration-time mortality response relationship of irritant and systemically acting vapors and gases. J. Hazard. Mater. 13:301–310.
- Tietz, N.W. 1986. Textbook of Clinical Chemistry. W.B. Saunders Co., Philadelphia, PA, p. 1821.
- Vernot, E.H., J.D.MacEwen, C.C.Haun and E.R.Kinkead. 1977. Acute toxicity and skin corrosion data for some organic and inorganic compounds and aqueous solutions. Toxicol. Appl. Pharmacol. 42:417–423. [PubMed: 595018]
- Vesey, C.J., P.V.Cole, P.J.Simpson. 1976. Cyanide and thiocyanate concentrations following sodium nitroprusside infusion in man. Br. J. Anaes. 48:651–660. [PubMed: 797397]
- Weedon, F.R, A.Hartzell, and C.Setterstrom. 1940. Toxicity of ammonia, chlorine, hydrogen cyanide, hydrogen sulfide and sulfur dioxide gases. V. Animals. Contrib. Boyce Thompson Inst. 11:365–385.
- Wexler, J., J.L.Whittenberger and P.R.Dumke. 1947. The effect of cyanide on the electrocardiogram of man. Amer. Heart J. 34:163–173. [PubMed: 20255260]
- Willhite, C.C. 1981. Malformations induced by inhalation of acetonitrile vapors in the golden hamster. Teratology 23:698.
- Willhite, C.C. 1982. Congenital malformations induced by laetrile. Science 215:1513–1515. [PubMed: 7063858]
- Willhite, C.C. and R.P.Smith. 1981. The role of cyanide liberation in the acute toxicity of aliphatic nitriles. Toxicol. Appl. Pharmacol. 59:589–602. [PubMed: 6267734]
- Willhite, C.C., V.H.Ferm, and R.P.Smith. 1982. Teratogenic effects of aliphatic nitriles. Teratology 23:317–323. [PubMed: 6266064]
- Yamanaka, S., Y.Takaku, and Y.Takaesu. 1991. Validity of salivary thiocyanate as an indicator of cyanide exposure from smoking. Bull. Tokyo Dental Coll. 32:157–163. [PubMed: 1668077]
Appendixes
APPENDIX A. TIME-SCALING CALCULATIONS FOR HYDROGEN CYANIDE
| Data: | |||
|---|---|---|---|
| Time (min) | Concentration (ppm) | Log time | Log concentration |
| 19 | 100 | 1.2788 | 2.0000 |
| 16 | 102 | 1.2041 | 2.0086 |
| 15 | 123 | 1.1761 | 2.0899 |
| 8 | 147 | 0.9031 | 2.1673 |
| 8 | 156 | 0.9031 | 2.1931 |
| Regression Output: | |
|---|---|
| Intercept | 2.6131 |
| Slope | −0.4769 |
| R Squared | 0.9142 |
| Correlation | −0.9561 |
| Degrees of Freedom | 3 |
| Observations | 5 |
| n=2.1 | |
| k=301326 | |
| Data: | |||
|---|---|---|---|
| Time (min) | Concentration (ppm) | Log time | Log concentration |
| 5 | 369 | 0.6990 | 2.5670 |
| 15 | 196 | 1.1761 | 2.2923 |
| 30 | 173 | 1.4771 | 2.2380 |
| 60 | 139 | 1.7782 | 2.1430 |
| Regression Output: | |
|---|---|
| Intercept | 2.8044 |
| Slope | −0.3854 |
| R Squared | 0.9490 |
| Correlation | −0.9742 |
| Degrees of Freedom | 2 |
| Observations | 4 |
| n=2.59 | |
| k=1.9E+07 | |
APPENDIX B. DERIVATION OF AEGL VALUES
Derivation of AEGL-1
| Key study: | Leeser et al. 1990 |
| Supporting studies: | El Ghawabi et al. 1975; Hardy et al. 1950; Grabois 1954; Maehlyand Swensson 1970; |
| Toxicity end point: | No adverse effect in healthy adult humans occupationally exposed at geometric mean concentration of ≤1 (range 0.01–3.3 ppm, personal samplers [up to 6 ppm, area samples]) or 5 ppm; mild headache in adult humans occupationally exposed at 8 ppm. The exposure duration was considered to be 8 h. |
| Uncertainty factor: | An uncertainty factor was not applied to the Leeser et al. (1990) 1-ppm concentration because it is the lowest NOAEL. A factor of 3 for intraspecies differences was applied to the supporting studies because no susceptible populations were identified. The uncertainty factor was applied to the 8-h 5 ppm and 8 ppm concentrations, which resulted in concentrations close to the 8-h 1-ppm concentration in the Leeser et al. (1990) study. |
| Scaling: | C3×t=k (conservative time-scaling relationship, because the relationship between concentration and exposure duration for the headache effect is unknown). An 8-h 1 ppm concentration was used as the starting point for time scaling. |
| Calculations: | (C3/uncertainty factors)×t=k (1 ppm)3×480 min=480 ppm3·min |
| 10-min AEGL-1: | (480 ppm3·min/10 min)1/3=3.6 ppm Because 3.6 ppm is above the highest exposure concentration in the Leeser et al. (1990) study, as measured by personal monitors, the 10-min value was set equal to the 30-min value. |
| 30-min AEGL-1: | (480 ppm3·min/30 min)1/3=2.5 ppm |
| 1-h AEGL-1: | (480 ppm3·min/60 min)1/3=2.0 ppm |
| 4-hour AEGL-1: | (480 ppm3·min/240 min)1/3=1.3 ppm |
| 8-hour AEGL-1: | 1.0 ppm |
Derivation of AEGL-2
| Key study: | Purser 1984 |
| Toxicity end point: | Slight central nervous system depression in monkeys inhaling 60 ppm for 30 min. |
| Scaling: | C2×t=k (this document; based on regression analysis of incapacitation and lethality data for the monkey) |
| Uncertainty factors: | 2 for interspecies 3 for intraspecies combined uncertainty factor of 6 |
| Calculations: | (C2/uncertainty factors)×t=k (60 ppm/6)2×30 min=3,000 ppm2·min |
| 10-min AEGL-2: | (3,000 ppm2·min/10 min)1/2=17 ppm |
| 30-min AEGL-2: | 60 ppm/6=10 ppm |
| 1-hour AEGL-2: | (3,000 ppm2·min/60 min)1/2=7.1 ppm |
| 4-hour AEGL-2: | (3,000 ppm2·min/240 min)1/2=3.5 ppm |
| 8-hour AEGL-2: | (3,000 ppm2·min/480 min)1/2=2.5 ppm |
Derivation of AEGL-3
| Key study: | E.I. du Pont de Nemours 1981 |
| Toxicity end point: | 15-min LC01 of 138 ppm in the rat 30-min LC01 of 127 ppm in the rat 1-h LC01 of 88 ppm in the rat LC01 derived by probit analysis |
| Scaling: | C2.6×t=k (this document; based on the E.I. du Pont de Nemours [1981] rat data set) |
| Uncertainty factors: | 2 for interspecies 3 for intraspecies combined uncertainty factor of 6 |
| Calculations: | (C2.6/uncertainty factors)×t=k (138 ppm/6)2.6×15 min=52,069.5 ppm2.6·min (127 ppm/6)2.6×30 min=83,911 ppm2.6·min (88 ppm/6)2.6×60 min=64,656.6 ppm2.6·min |
| 10-min AEGL-3: | (52,069.5 ppm2.6·min/10 min)1/2.6=27 ppm |
| 30-min AEGL-1: | 127 ppm/6=21 ppm |
| 1-h AEGL-1: | 88 ppm/6=15 ppm |
| 4-h AEGL-1: | (64,656.6 ppm2.6·min/240 min)1/2.6=8.6 ppm |
| 8-h AEGL-1: | (64,656.6 ppm2.6·min/480 min)1/2.6=6.6 ppm |
APPENDIX C. DERIVATION SUMMARY FOR ACUTE EXPOSURE GUIDELINE LEVELS FOR HYDROGEN CYANIDE (CAS No. 74–90–8)
| AEGL-1 | ||||
|---|---|---|---|---|
| 10 min | 30 min | 1 h | 4 h | 8 h |
| 2.5 ppm | 2.5 ppm | 2.0 ppm | 1.3 ppm | 1.0 ppm |
| Key reference: | Leeser, J.E., J.A.Tomenson, and D.D.Bryson. 1990. A cross-sectional study of the health of cyanide salt production workers. Report No. OHS/R/2, ICI Central Toxicology Laboratory, Alderley Park, Maccles field, Cheshire, U.K. | |||
| Supporting references: |
| |||
| Test Species/Strain/Number: Occupational exposures/63 employees, mean age 44.7 (Leeser et al. 1990) Occupational exposures/36 workers (El Ghawabi et al. 1975) Occupational exposures/five factories (Grabois 1954) Occupational exposures/94 workers (Maehly and Swensson 1970) Occupational exposures/factories (Hardy et al. 1950) | ||||
| Exposure Route/Concentrations/Durations: Inhalation/geometric mean exposure of ≤1 ppm (range, 0.01–3.3 ppm; personal samplers), up to 6 ppm (area samples)/mean service years, 16.5 (Leeser et al. 1990); Inhalation/average exposure 8 ppm/5–15 y (El Ghawabi et al. 1975); Inhalation/5 ppm/unknown/(Grabois 1954; Maehly and Swensson 1970; Hardy et al. 1950). | ||||
| Effects: No exposure related adverse symptoms or health effects (surveys and medical examinations taken in spring and fall of year) (Leeser et al. 1990); mild headache, other symptoms (El Ghawabi et al. 1975); no effects reported (Grabois 1954; Maehly and Swensson 1970; Hardy et al. 1950). | ||||
| End point/Concentration/Rationale: 1 ppm from the Leeser (1990) study; 8 ppm from the El Ghawabi et al. (1975) study; or 5 ppm from the Hardy et al. (1950), Grabois (1954), and Maehly and Swensson (1970) studies were considered no-adverse-effect to mild effect concentrations for an 8-h work day. The NRC adjusted the chronic 8 ppm value of El Ghawabi et al. (1975) to a 1-h exposure for healthy adults. | ||||
| Uncertainty factors/Rationale: Total uncertainty factor: 3 | ||||
| Interspecies: | Not applicable | |||
| Intraspecies: | An uncertainty factor was not applied to the Leeser et al. (1990) 1 ppm concentration, as it is the lowest NOAEL. A factor of 3 was applied to the supporting studies as no specific susceptible populations were identified in monitoring studies or during the clinical use of nitroprusside solutions to control hypertension. The detoxifying enzyme rhodanese is present in all individuals including newborns. Application of the uncertainty factor to the El Ghawabi et al. (1975; as adjusted by the NRC) and Grabois (1954) data results in a value close to the 8-h 1 ppm concentration in the Leeser et al. (1990) study. | |||
| Modifying factor: Not applicable | ||||
| Animal to human dosimetric adjustment: Not applicable | ||||
| Time scaling: Because of the long-term exposure duration of the key studies, the conservative time-scaling value of n=3 (k=480 ppm3·min) was applied when scaling to shorter exposure durations. The starting point for time scaling was an 8-h concentration at 1 ppm. | ||||
| Data adequacy: The preponderance of data from the key studies support an 8-h no-effect concentration of 1 ppm. The Leeser et al. (1990) study encompassed subjective symptoms as well as extensive medical examinations. The occupational monitoring study of El Ghawabi et al. (1975), in which it is believed that workers inhaling a mean concentration of 8 ppm may suffer mild headaches, supports the safety of the derived values. The values are also supported by a NIOSH (1976) report in which 5 ppm was identified as a no-effect concentration in the Grabois et al. (1954) occupational study. Additional monitoring studies support the values. | ||||
| AEGL-2 | ||||
| 10 min | 30 min | 1 h | 4 h | 8 h |
| 17 ppm | 10 ppm | 7.1 ppm | 3.5 ppm | 2.5 ppm |
| Key references: |
| |||
| Test species/Strain/Sex/Number: Cynomolgus monkeys, 4 per exposure group (gender not stated) | ||||
| Exposure route/Concentrations/Durations: Inhalation, 60, 100, 102, 123, 147, or 156 ppm for 30 min | ||||
| Effects: (30-min exposures) | ||||
| 60 ppm | increased respiratory minute volume and slight changes in EEGs near end of exposure | |||
| 100 ppm | incapacitation (semi-conscious state) in 19 min | |||
| 102 ppm | incapacitation in 16 min | |||
| 123 ppm | incapacitation in 15 min | |||
| 147 ppm | incapacitation in 8 min | |||
| 156 ppm | incapacitation in 8 min | |||
| End point/Concentration/Rationale: The 30-min exposure to 60 ppm, a NOAEL, was chosen because the next higher tested concentration, 100 ppm, resulted in incapacitation within the 30-min exposure period. | ||||
| Uncertainty factors/Rationale: Total uncertainty factor: 6 | ||||
| Interspecies: | 2—The monkey is an appropriate model for humans, the small size and higher respiratory rate of the monkey may result in more rapid uptake and greater sensitivity than in humans. | |||
| Intraspecies: | 3—No specific susceptible populations were identified during monitoring studies or during the clinical use of nitroprusside solutions to control hypertension. The detoxifying enzyme rhodanese is present in all individuals, including newborns. | |||
| Modifying factor: Not applicable | ||||
| Animal to human dosimetric adjustment: Insufficient data. | ||||
| Time scaling: | Cn×t=k, where n=2 and k=3,000 ppm2·min on the basis of regression analysis of time-concentration relationships for both incapacitation times of 8 to 19 min and lethality data (3–60 min) for the monkey. | |||
| Data Adequacy: Although human data are limited to primarily occupational monitoring studies, the data base on animal studies is good. The test atmosphere in the key study was supplied via a face mask to the restrained test subjects; restrained animals have been shown to be more sensitive than unrestrained animals to inhaled toxicants. Relative species sensitivity to inhaled HCN may be related to breathing rate. Compared to rodents, the slower breathing rate of humans and monkeys may make them less sensitive to the effects of HCN. The following two supporting studies were located:
| ||||
| AEGL-3 | ||||
| 10 min | 30 min | 1 h | 4 h | 8 h |
| 27 ppm | 21 ppm | 15 ppm | 8.6 ppm | 6.6 ppm |
| Key reference: | E.I. du Pont de Nemours and Company 1981. Inhalation toxicity of common combustion gases. Haskell Laboratory Report No. 238–81. Haskell Laboratory, Newark, DE | |||
| Test species/Strain/Sex/Number: Crl:CD male rats, 10/exposure group | ||||
| Exposure route/Concentrations/Durations: Inhalation 273, 328, 340, 353, 441, 493, or 508 ppm for 5 min 110, 175, 188, 204, 230, 251, 283, or 403 ppm for 15 min 128, 149, 160, 183, 222, or 306 ppm for 30 min 76, 107, 154, 183, or 222 ppm for 60 min | ||||
| Effects (LC01 values were calculated by Haskell Laboratory using probit analysis): | ||||
| 5-min LC01: | 283 ppm | |||
| 15-min LC01: | 138 ppm | |||
| 30-min LC01: | 127 ppm | |||
| 60-min LC01: | 88 ppm | |||
| End point/Concentration/Rationale: The LC01, the threshold for lethality, was used as the basis for the derivation of the AEGL-3. The 15-min LC01 was used to calculate the 10-min value; the 30-min LC01 was used for the 30-min value; and the 60-min LC01 was used to derive the 1-, 4-, and 8-h AEGL-3 values. | ||||
| Uncertainty factors/Rationale: Total uncertainty factor: 6 | ||||
| Interspecies: | 2—LC50 values for the same exposure durations for several species (rat, mouse, and rabbit) were within a factor of approximately 1.5 of each other. Based on relative respiration rates, humans are expected to be less sensitive than rodents. The mechanism is the same for all species. | |||
| Intraspecies: | 3—No specific susceptible populations were identified during monitoring studies or during the clinical use of nitroprusside solutions to control hypertension. The detoxifying enzyme rhodanese is present in all individuals, including newborns. | |||
| Modifying factor: Not applicable | ||||
| Animal to human dosimetric adjustment: Insufficient data. | ||||
| Time scaling: | Cn×t=k where n=2.6 was derived from empirical data and used in a regression analysis of time-concentration relationships for rat LC50 values conducted at time periods of 5, 15, 30, and 60 min in the key study. However, the 15-, 30-, and 60-min values were calculated directly from the empirical (LC01) data. The k value of 52,069.5 ppm2.6·min, based on the 15-min LC01, was used for the 10-min value and the k value of 64,656.6 ppm2.6·min, based on the 1-h LC01, was used for the 4- and 8-h AEGL-3 values. | |||
| Data adequacy: The study was well conducted. The HCN concentrations were continuously monitored using infrared spectrophotometry and validated by gas chromatography. One supporting study was located: exposure of rats to 30 ppm for 24 hours resulted in lung congestion but no deaths. Use of a total uncertainty factor of 6 and extrapolation across time to 30 minutes results in a 30-minute AEGL-3 of 22 ppm which is similar to the derived value of 21 ppm. Sumber : sumber ilmiah dalam net | ||||
Tidak ada komentar:
Posting Komentar